Code
source("03.script/00_packages.R")source("03.script/00_packages.R")Les données proviennent d’un tableau Excel (.xlsx).
Package utilisé
- readxl
OUTPUT
- DATA [402 x 3824]: dataframe contenant l’ensemble des données floristiques et des métadonnées.
Les données brutes sont coupées pour ne garder que les relevés de 2021 (DATA_21), correspondant aux derniers relevés en date. A cette date, les relevés ont également été effectué en Charente-Maritime, ceux-ci ont aussi été enlevés.
Packages utilisés
- dplyr
- stringr
source("03.script/02_cleaning.R")OUTPUT
- DATA_21 [401 x 454] : dataframe contenant les métadonnées et les relevés floristiques de 2021 et uniquement de la Gironde et des Landes.
- METADATA [453 x 64] : contient les variables environnementales et logistiques.
- FLORE [453 x 343] : contient les valeurs de coefficient de Braun-Blanquet pour chaque relevé et chaque espèce.
source("03.script/03_functions.R")Informations descriptives du jeu de données. Nombre d’espèces recensées en 2021 et leur occurence dans les relevés et les transects.
Package utilisé
- dplyr
species_cols <- !colnames(FLORE) %in% c("transect_numb", "REL") # Pour ne convertir que les esp
# Richesse spécifique 2021
richesse_all <- sum(colSums(FLORE[, species_cols] != "", na.rm = TRUE) > 0) # 109
# Liste des espèces recensées (présentes au moins une fois)
esp_recensees <- colnames(FLORE[, species_cols])[
colSums(FLORE[, species_cols] != "", na.rm = TRUE) > 0
]
# Analyses exploratoires espèce-centrée
Recensement <- tibble(
Espèce = esp_recensees,
Nb_relevés = colSums(FLORE[, esp_recensees] != "", na.rm = TRUE)
) %>% # espèces + nombre de relevés où elles sont présentes
mutate(
Nb_Transects = sapply(esp_recensees, function(sp) {
length(unique(FLORE$transect_numb[FLORE[[sp]] != ""]))
}) -
1
) %>% # nombre de transects où chaque espèce est présente
arrange(desc(Nb_relevés)) # les espèces les plus fréquentes au débutOUTPUT
- richesse_all : richesse spécifique sur l’ensemble des données 2021.
- esp_recensees : liste des espèces présentes au moins une fois dans les données.
- Recensement : pour chaque espèce, nombre de relevés et le nombre de transects dans lesquelles elle apparaît.
facies <- METADATA %>%
filter(!is.na(Type_habitat)) %>%
count(Type_habitat, sort = TRUE)chisq.test(facies$n)
Chi-squared test for given probabilities
data: facies$n
X-squared = 1137.8, df = 16, p-value < 2.2e-16Le coefficient de Braun-Blanquet est composé de classes de présence correspondant à des fréquences relatives (%). Pour analyser ces données, les valeurs d’abondance-dominance selon Braun-Blanquet (1932) ont été remplacées par le recouvrement moyen (%), comme présenté par Dufrêne (1998 in Meddour 2011) via la fonction bb_to_cover.
Package utilisé
- dplyr
#### FLORE_cover ----
FLORE_cover <- apply(FLORE[, species_cols], 2, bb_to_cover) # applique la fonction à toutes les colonnes espèces de FLORE -> matrice % recouvrement
FLORE_cover <- as.data.frame(FLORE_cover) # matrce -> dataframe
FLORE_cover[is.na(FLORE_cover)] <- 0 # Remplace les NA par 0 pour calculer Shannon
FLORE_cover$REL <- FLORE$REL # ajoute colone REL depuis FLORE
FLORE_cover$transect_numb <- FLORE$transect_numb # ajoute colonne transect depuis FLORE
FLORE_cover <- FLORE_cover %>%
select(REL, transect_numb, everything()) # Met le numéro du relevé et du transect en premières colonnes
##### REL_to_transect ----
# REL_to_transect permet de garder un tableau de conversion
REL_to_transect <- FLORE %>%
select(REL, transect_numb) %>%
distinct() # table de jointureOUTPUT
- bb_to_cover : fonction qui convertit le coefficient de Braun-Blanquet (Braun-Blanquet, 1932) en % de recouvrement moyen (Dufrêne 1998 in Meddour 2011).
- species_cols : vecteur logique pour séparer les colonnes d’esp avec les autres colonnes. Permet de sélectionner uniquement les colonnes d’espèces.
- FLORE_cover [453 x 343] : dataframe avec le recouvrement moyen (%) des espèces en fontion des relevés.
- REL_to_transect [453 x 2] : dataframe permettat la mise en relation des relevés avec le transect correspondant.
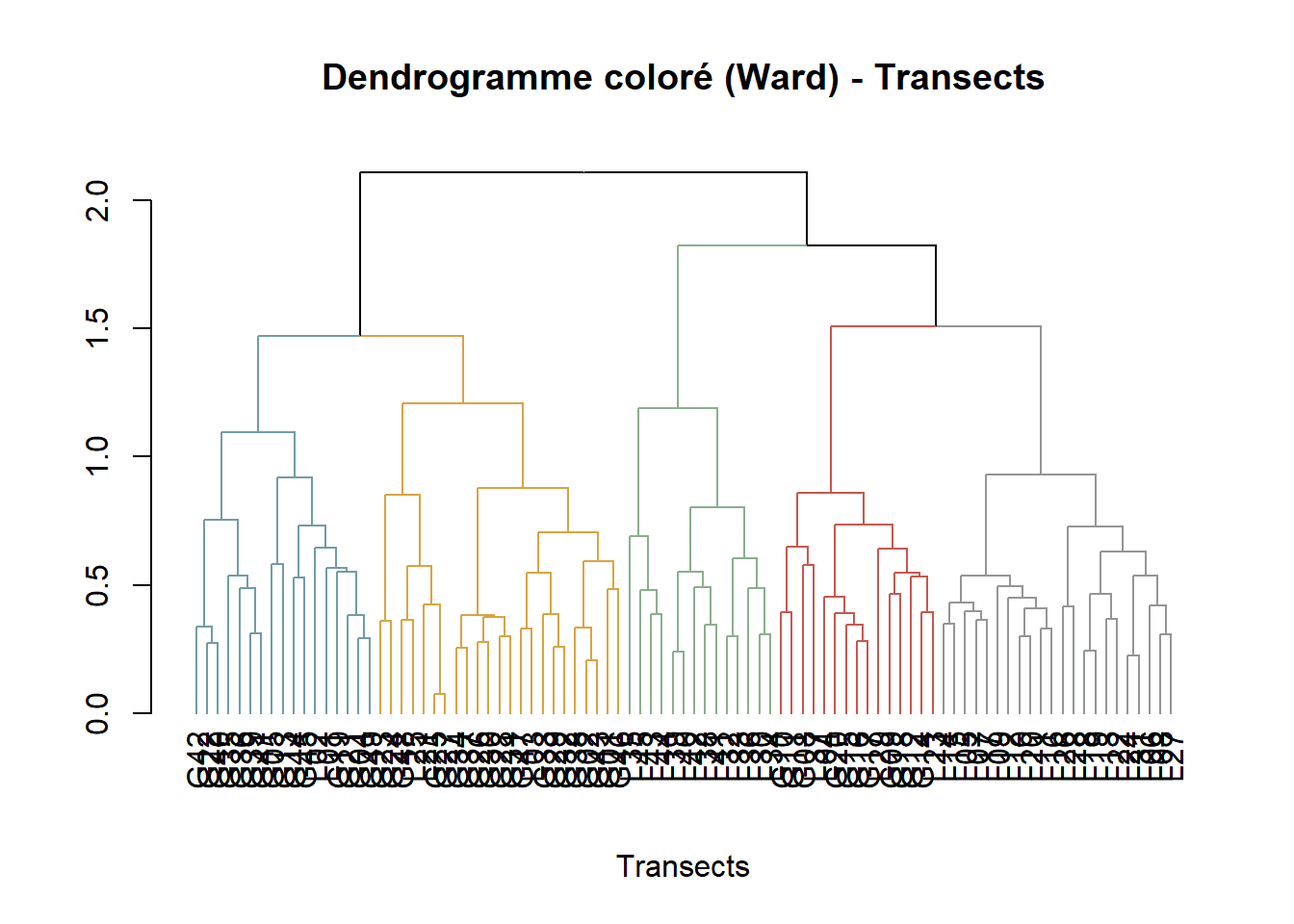
Utilisation de dataframe FLORE_cover_esp transformé en log -réduit l’effet des dominants- afin d’obtenir une matrice de distance avec la méthode de Bray-Curtis (Bray & Curtis, 1957). La méthode Bray-Curtis est utilisée pour mesurer les différences en abondances entre relevés. La métrique de distance Bray-Crutis est non-métrique, c’est pourquoi elle ne peut pas être utilisé avec l’algorithme de regroupe de Ward qui nécessite une distance métrique. Pour cela, les résultats Bray-Curtis sont transformés en racine carrée. Les clusters sont créés via la méthode de Ward.D2 qui maximise la variance intra-groupe.
Packages utilisés
- vegdist
- dendextend
#### Préparation data ----
FLORE_cover_esp <- FLORE_cover[, species_cols]
FLORE_cover_esp <- FLORE_cover_esp[rowSums(FLORE_cover_esp, na.rm = TRUE) > 0, ]
FLORE_cover_esp <- FLORE_cover_esp[, colSums(FLORE_cover_esp, na.rm = TRUE) > 0]
FLORE_cover_esp_log <- log1p(FLORE_cover[, species_cols]) # Réduction des effets des dominants
rownames(FLORE_cover_esp_log) <- FLORE_cover$REL
FLORE_cover_esp_log <- FLORE_cover_esp_log[
rowSums(FLORE_cover_esp_log, na.rm = TRUE) > 0,
]
FLORE_cover_esp_log <- FLORE_cover_esp_log[,
colSums(FLORE_cover_esp_log, na.rm = TRUE) > 0
]
#### Dendrogramme ----
dist_mat <- vegdist(FLORE_cover_esp_log, method = "bray") # création d'une matrie de distance avec la méthode de Bray-Curtis
dist_mat_sqrt <- sqrt(dist_mat) # rend la matrice de distance de BC en racine carrée
clust <- hclust(dist_mat_sqrt, method = "ward.D2") # Maximise la variance intra-groupe
groups <- cutree(clust, k = 4) # Décidé par observation
# DF REL + groupes
rel_groups <- tibble(
REL = names(groups),
Groupe = as.factor(groups)
)
# Joinure avec METADATA (X, Y)
rel_groups <- rel_groups %>%
left_join(
METADATA %>% select(REL, transect_numb, X, Y, Type_habitat),
by = "REL"
) %>%
arrange(Groupe, REL)
group_palette <- c(
"1" = "#709CA7",
"2" = "#D9A441",
"3" = "#8BAF8D",
"4" = "#C8574D",
"5" = "#939597"
)L’ANOSIM (test non-paramétrique) teste si les rangs dissimilarités entre-groupes > intra-groupes.
Package utilisé
- vegan
anosim_res <- anosim(FLORE_cover_esp_log, groups, permutations = 999) # "999 permutations" permet d'être précis et rapide
Call:
anosim(x = FLORE_cover_esp_log, grouping = groups, permutations = 999)
Dissimilarity: bray
ANOSIM statistic R: 0.5005
Significance: 0.001
Permutation: free
Number of permutations: 999L’ANOSIM confirme une différence significative entre les 4 groupes de relevés (p_value = 0.001). Les rangs de dissimilartés Bray-Curtis entre groupe sont supérieurs aux rangs intra-groupes, avec une séparation modérée (R = 0.44) donc garde un certain gradient.
Le SIMPER (Clarke, 1993) identifie quelles espèces expliquent les différences entre les groupes, espèces indicatrices.
Package utilisé
- vegan
simper_res <- simper(FLORE_cover_esp, factor(groups), permutations = 99)
characteristic_species <- top5_all(simper_res, top_n = 5)
df_simper <- map_dfr(
1:6,
~ {
x <- summary(simper_res, ordered = TRUE)[[.x]] %>%
filter(p <= 0.01 & ratio > 1.0) %>%
rownames_to_column("Espece")
if (nrow(x) > 0) {
mutate(x, Paire = names(summary(simper_res, ordered = TRUE))[.x])
}
},
.id = "Rang"
) %>%
select(Paire, Espece, average, ratio, p)$`1`
x
Ammophila arenaria Corynephorus canescens
3 3
Helichrysum stoechas Galium arenarium
3 2
Artemisia campestris ssp maritima Carex arenaria
1 1
Elytrigia juncea Euphorbia paralias
1 1
$`2`
x
Ammophila arenaria Galium arenarium
3 3
Eryngium maritimum Euphorbia paralias
2 2
Artemisia campestris ssp maritima Convolvulus soldanella
1 1
Corynephorus canescens Elytrigia juncea
1 1
Helichrysum stoechas
1
$`3`
x
Ammophila arenaria Artemisia campestris ssp maritima
3 3
Eryngium maritimum Galium arenarium
2 2
Carex arenaria Corynephorus canescens
1 1
Elytrigia juncea Euphorbia paralias
1 1
Helichrysum stoechas
1
$`4`
x
Ammophila arenaria Elytrigia juncea
3 3
Galium arenarium Eryngium maritimum
3 2
Artemisia campestris ssp maritima Convolvulus soldanella
1 1
Corynephorus canescens Helichrysum stoechas
1 1 id_cols <- c("REL", "transect_numb")
species_cols_name <- setdiff(names(FLORE_cover), id_cols)
df_species <- FLORE_cover %>%
select(all_of(c("REL", species_cols_name))) %>%
pivot_longer(
all_of(species_cols_name),
names_to = "Espece",
values_to = "Cover"
) %>%
filter(Cover > 0)
df_species <- df_species %>%
left_join(rel_groups %>% select(REL, Groupe), by = "REL")
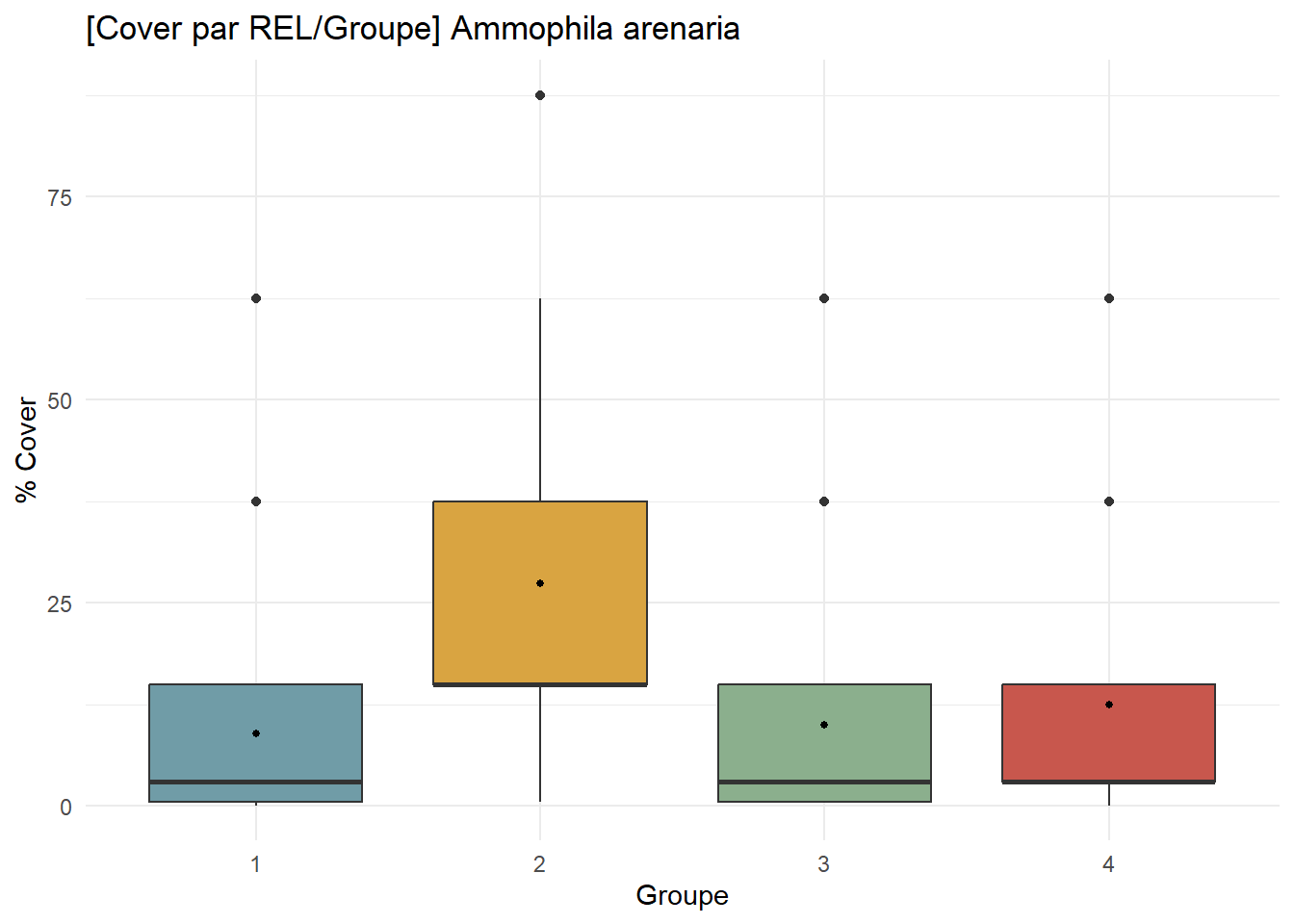
# Ammophila arenaria
AA_contribution <- ggplot(
df_species %>% filter(Espece == "Ammophila arenaria"), # sélectionne uniquement Ammophila
aes(x = Groupe, y = Cover, fill = Groupe)
) +
geom_boxplot() +
stat_summary(fun = mean, geom = "point", size = 1, fill = "white") +
scale_fill_manual(values = group_palette) +
labs(
title = "[Cover par REL/Groupe] Ammophila arenaria",
x = "Groupe",
y = "% Cover"
) +
theme_minimal() +
theme(legend.position = "none")
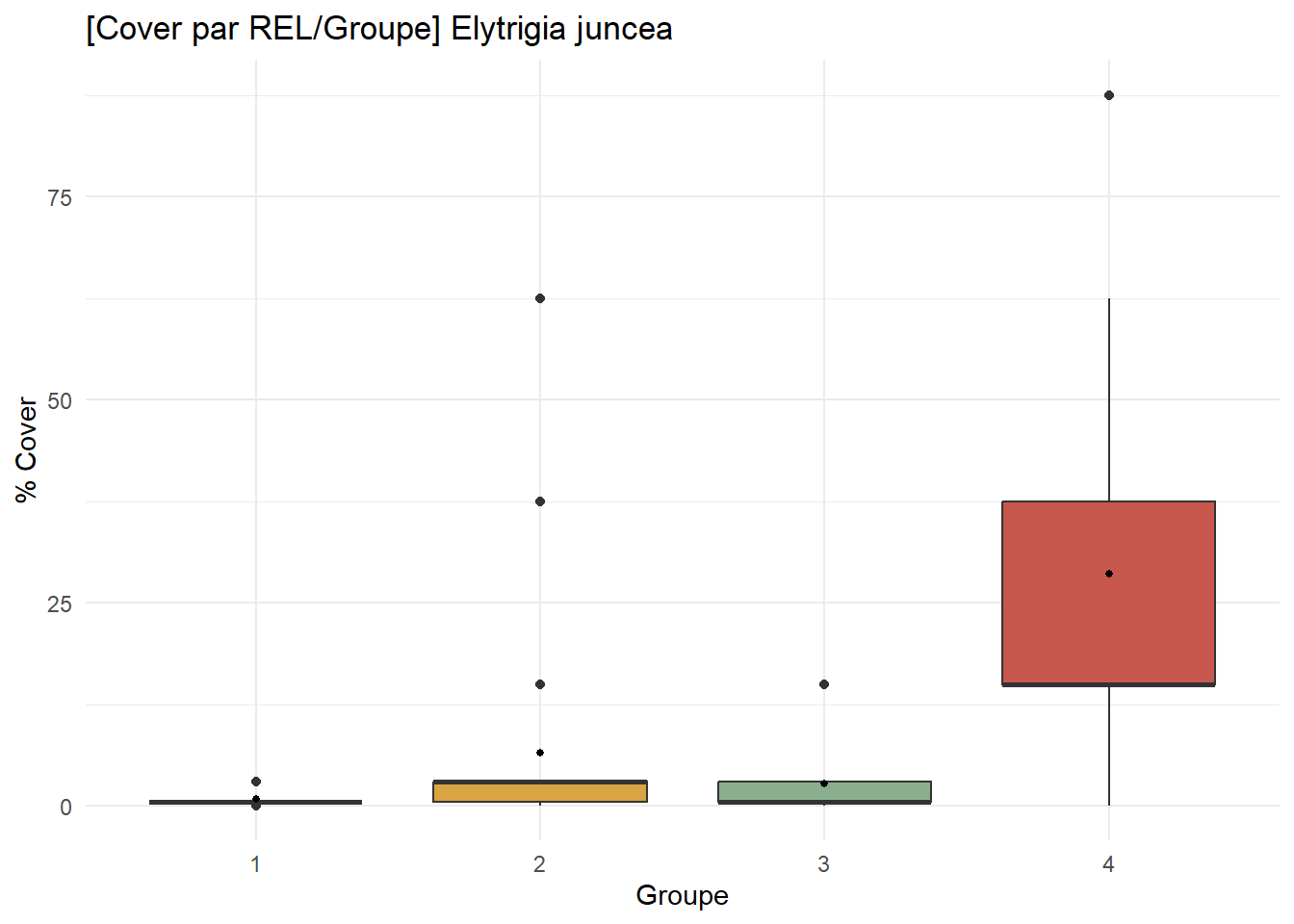
# Elytrigia juncea
EJ_contribution <- ggplot(
df_species %>% filter(Espece == "Elytrigia juncea"),
aes(x = Groupe, y = Cover, fill = Groupe)
) +
geom_boxplot() +
stat_summary(fun = mean, geom = "point", size = 1, fill = "white") +
scale_fill_manual(values = group_palette) +
labs(
title = "[Cover par REL/Groupe] Elytrigia juncea",
x = "Groupe",
y = "% Cover"
) +
theme_minimal() +
theme(legend.position = "none")
# Helichrysum stoechas
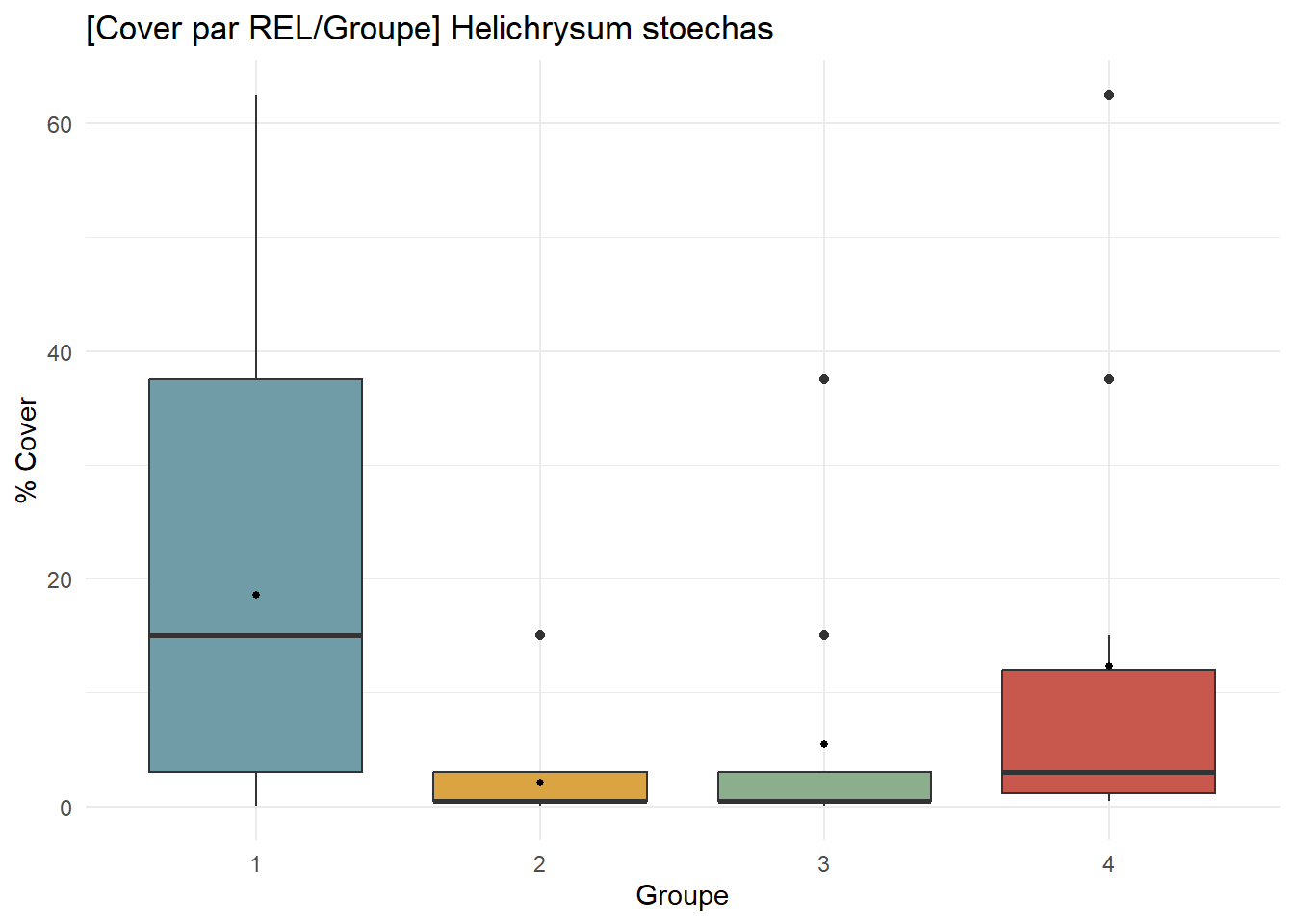
HS_contribution <- ggplot(
df_species %>% filter(Espece == "Helichrysum stoechas"),
aes(x = Groupe, y = Cover, fill = Groupe)
) +
geom_boxplot() +
stat_summary(fun = mean, geom = "point", size = 1, fill = "white") +
scale_fill_manual(values = group_palette) +
labs(
title = "[Cover par REL/Groupe] Helichrysum stoechas",
x = "Groupe",
y = "% Cover"
) +
theme_minimal() +
theme(legend.position = "none")
# Corynephorus canescens
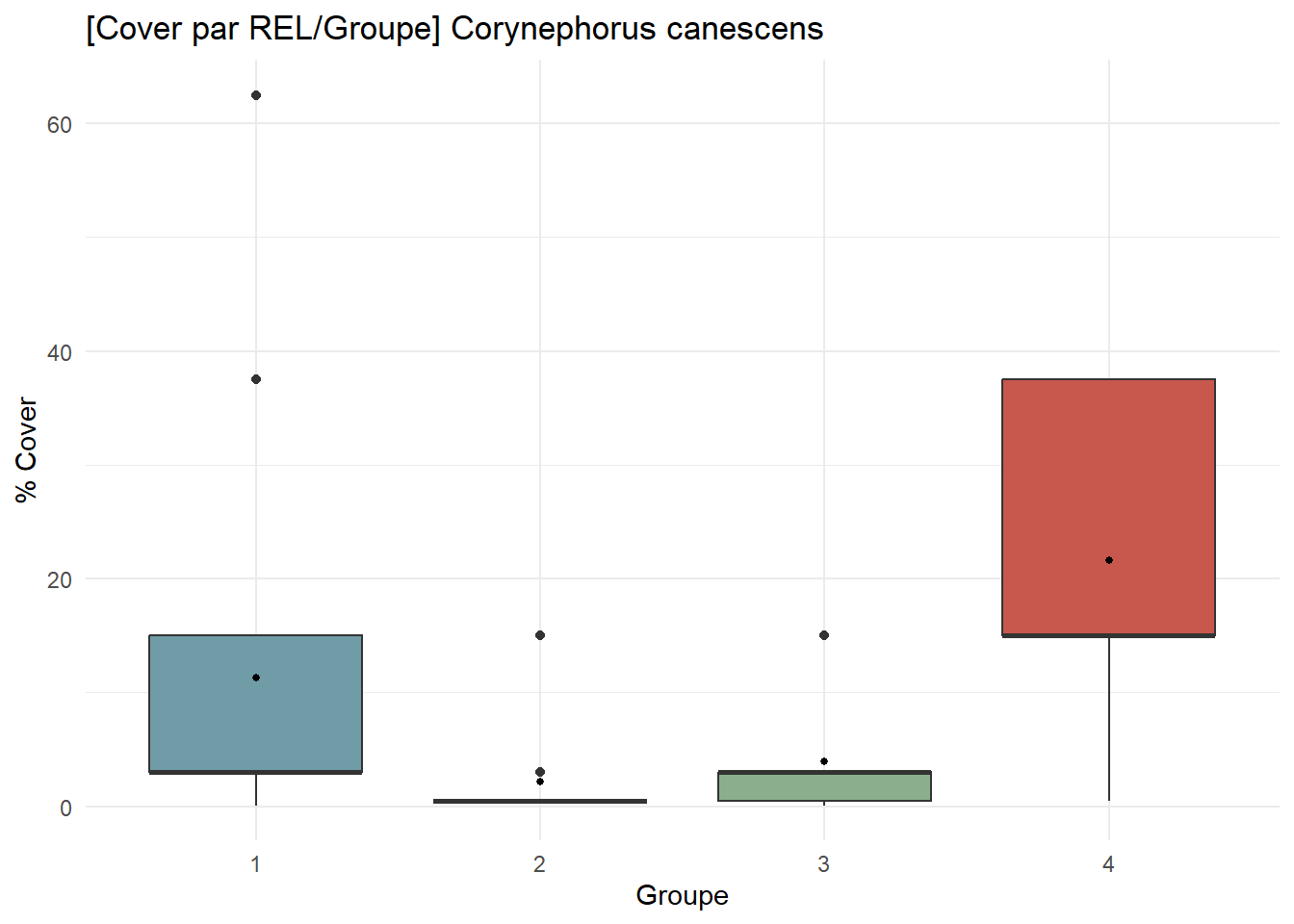
CC_contribution <- ggplot(
df_species %>% filter(Espece == "Corynephorus canescens"),
aes(x = Groupe, y = Cover, fill = Groupe)
) +
geom_boxplot() +
stat_summary(fun = mean, geom = "point", size = 1, fill = "white") +
scale_fill_manual(values = group_palette) +
labs(
title = "[Cover par REL/Groupe] Corynephorus canescens",
x = "Groupe",
y = "% Cover"
) +
theme_minimal() +
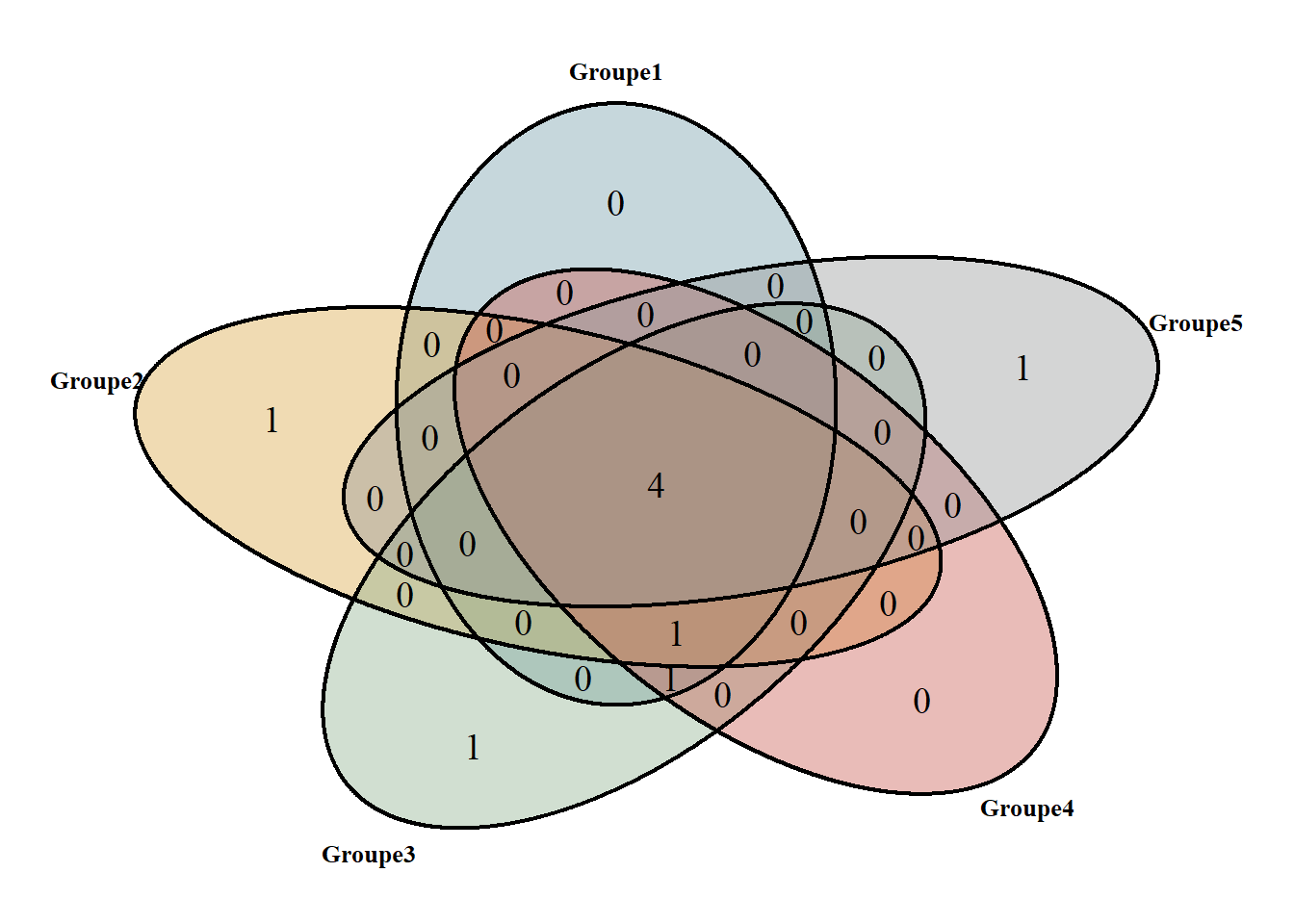
theme(legend.position = "none")Le diagramme de Venn permet de visualiser les résultats du SIMPER.
Package utilisé
- VennDiagram
#### Extraction espèces inditrices ----
extract_species_filtered <- function(simper_obj, top_n = 5, min_freq = 2) {
species_list <- list()
for (comp in names(simper_obj)) {
df <- as.data.frame(simper_obj[[comp]])
df <- df[order(-df$average), ] # tri décroissant
top_species <- head(rownames(df), top_n) # top 5
groups <- strsplit(comp, "_")[[1]]
for (g in groups) {
species_list[[g]] <- c(species_list[[g]], top_species)
}
}
lapply(species_list, function(x) {
freq <- sort(table(x), decreasing = TRUE)
names(freq[freq >= min_freq])
})
}
filtered_species <- extract_species_filtered(
simper_res,
top_n = 5,
min_freq = 2
)
#### Visualisation ----
venn.plot <- venn.diagram(
x = list(
Groupe1 = filtered_species[["1"]],
Groupe2 = filtered_species[["2"]],
Groupe3 = filtered_species[["3"]],
Groupe4 = filtered_species[["4"]]
),
filename = NULL,
fill = group_palette[as.character(1:4)],
alpha = 0.4,
cex = 1.2,
cat.cex = 0.8,
cat.fontface = "bold",
margin = 0.1
)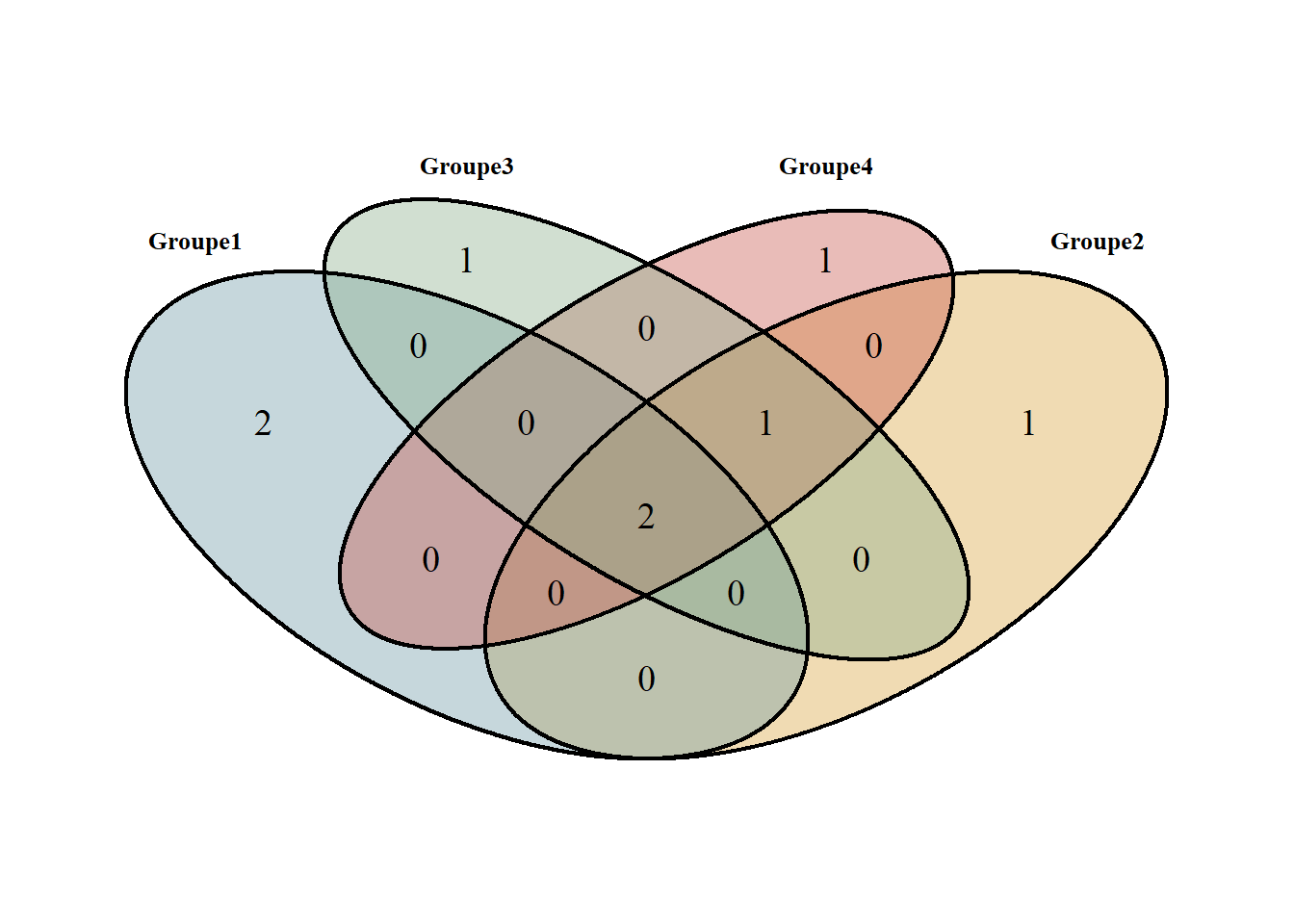
2 espèces dans tous les groupes (Ammophila) Télécharger ce graphique
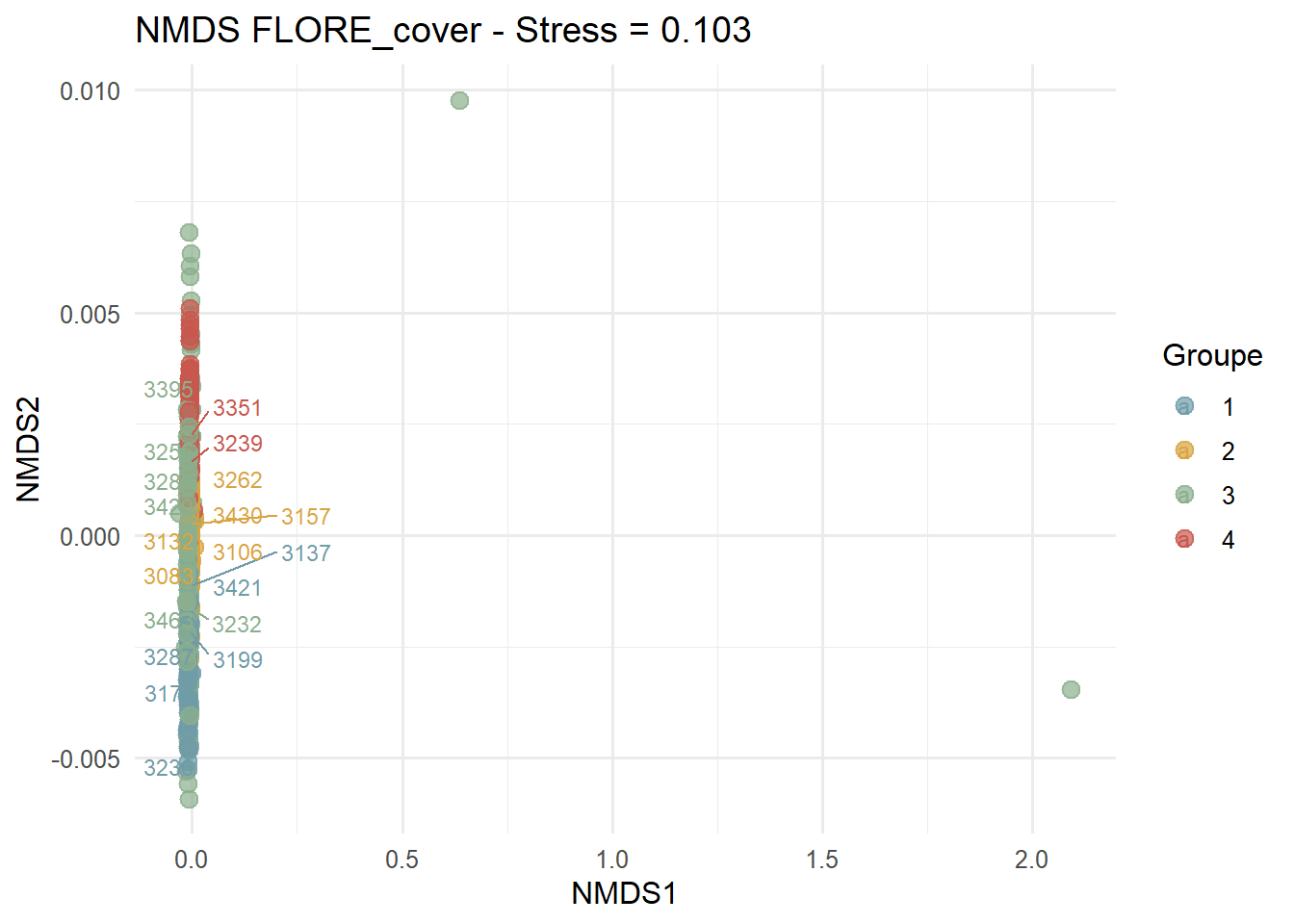
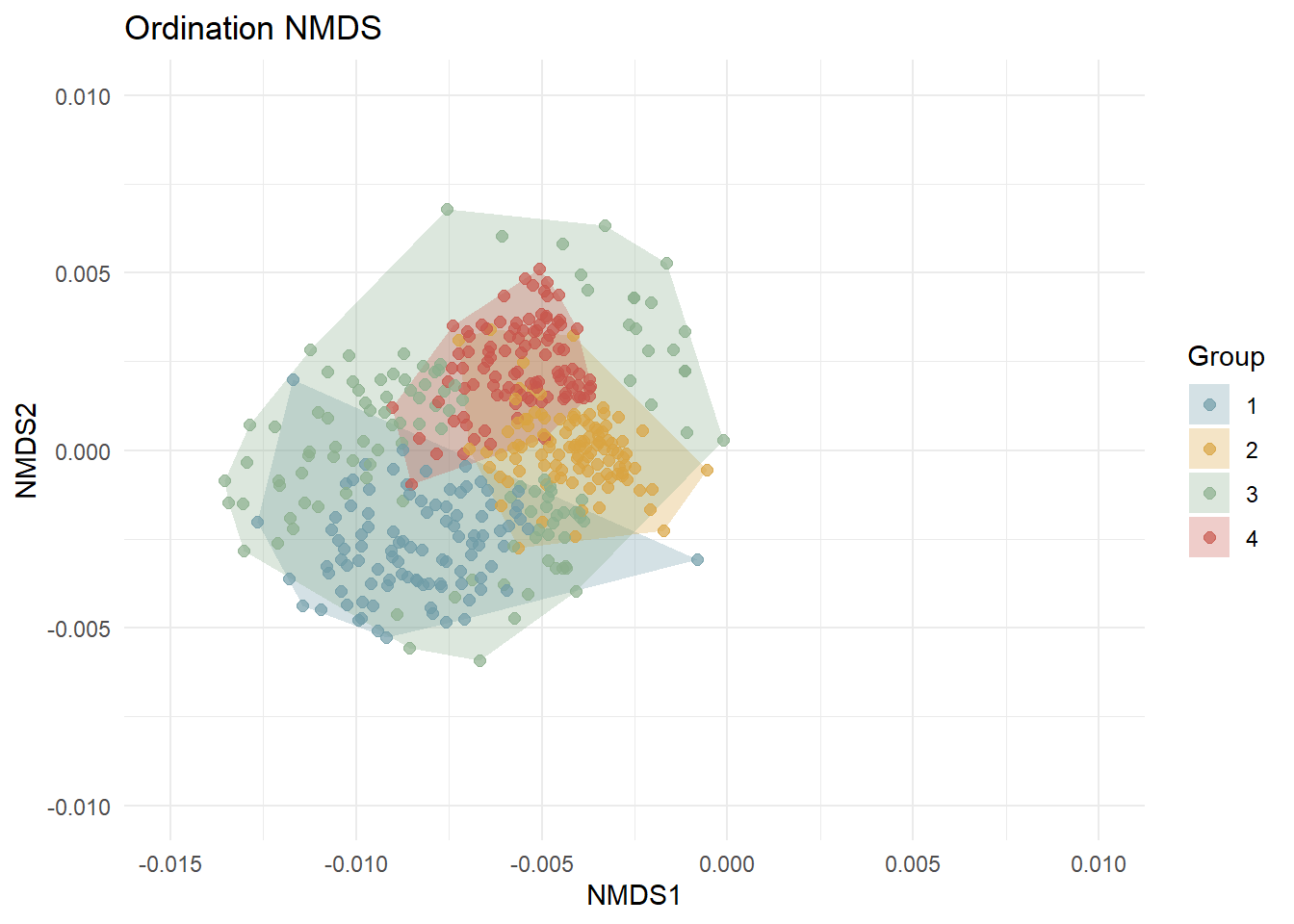
Analyses multivariées avec un cadrage multidimentionnel (ici, 2 dimensions) non-paramétrique. Calcul des distances avec la méthode de Bray-Curtis, 100 essais. La détection des outliers se fait avec la méthode IQR (écart interquartile) de Tuckey (1977 in Lin et al., 2025). Tous les points de données qui se trouvent au-delà de 1,5 fois l’IQR en dessous du premier quartile ou au-dessus du troisième quartile sont exclus.
# 1. nMDS ordination
nmds <- metaMDS(FLORE_cover_esp_log, distance = "bray", k = 2, try = 100)[1] "Stress = 0.103"# 2. Coordonées REL
nmds_points <- as.data.frame(scores(nmds, display = "sites"))
nmds_points$REL <- rownames(nmds_points)
# 3. Jointure groupes
nmds_points <- nmds_points %>%
left_join(
data.frame(REL = names(groups), Group = as.character(groups)),
by = "REL"
)
nmds_points <- nmds_points %>%
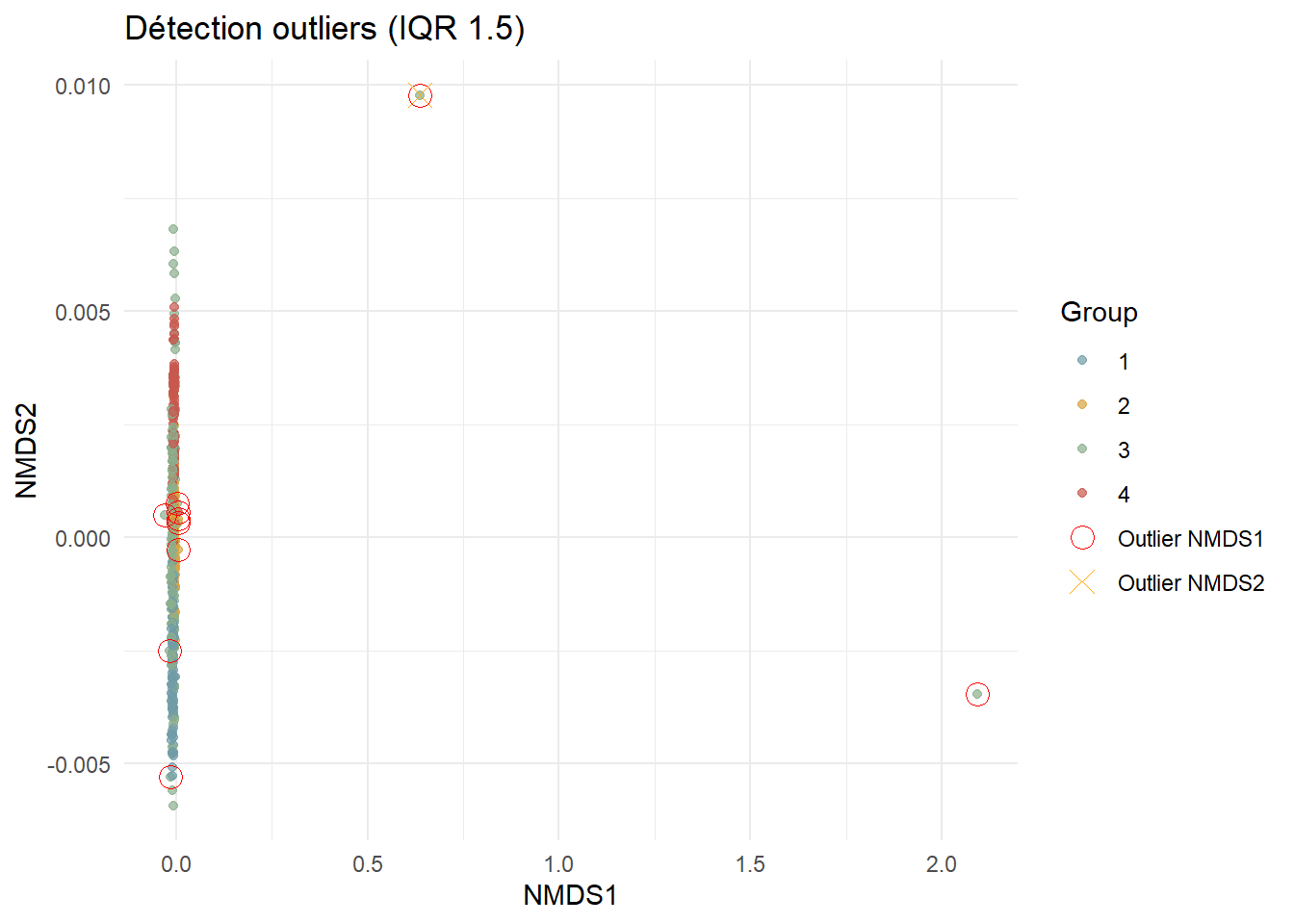
filter(!is.na(NMDS1), !is.na(NMDS2))La méthode IQR est utilisée pour connaître les outliers.
# Détection des outliers
detect_outliers <- function(data, var) {
Q1 <- quantile(data[[var]], 0.25)
Q3 <- quantile(data[[var]], 0.75)
IQR_val <- Q3 - Q1
lower <- Q1 - 1.5 * IQR_val
upper <- Q3 + 1.5 * IQR_val
outliers <- data[data[[var]] < lower | data[[var]] > upper, ]
list(outliers = outliers, bounds = c(lower, upper))
}
outliers1 <- detect_outliers(nmds_points, "NMDS1")
outliers2 <- detect_outliers(nmds_points, "NMDS2")nmds_clean <- nmds_points %>%
filter(
!row_number() %in% row.names(outliers1$outliers),
!row_number() %in% row.names(outliers2$outliers)
)
# 5. Enveloppes
hulls <- nmds_clean %>%
group_by(Group) %>%
filter(n() >= 3) %>%
slice(chull(NMDS1, NMDS2)) %>%
ungroup()
# 6. Visualisation + limites
NMDS_clean <- ggplot(nmds_clean, aes(x = NMDS1, y = NMDS2, color = Group)) +
geom_polygon(
data = hulls,
aes(fill = Group),
alpha = 0.3,
color = 'NA',
linewidth = 0.5
) +
geom_point(size = 2, alpha = 0.7) +
xlim(-0.015, 0.01) +
ylim(-0.01, 0.01) + # limites manuelles
scale_color_manual(values = group_palette) +
scale_fill_manual(values = group_palette) +
theme_minimal() +
labs(title = "Ordination NMDS", x = "NMDS1", y = "NMDS2")Packages uilisés
- pheatmap
- RColorBrewer
#### Préparation data ----
annotation_col <- data.frame(Cluster = factor(groups))
rownames(annotation_col) <- names(groups)
freq <- colSums(FLORE_cover_esp_log > 0, na.rm = TRUE)
cols_heat <- colorRampPalette(c("#FFFFFF", "#CE6A6B"))(100)
ann_colors <- list(Cluster = group_palette)
#### Visualisation ----
heatmap <- pheatmap(
t(FLORE_cover_esp_log),
color = cols_heat,
cluster_rows = FALSE,
cluster_cols = clust,
annotation_col = annotation_col,
fontsize = 3,
annotation_colors = ann_colors,
show_rownames = TRUE, # Afficher nom espèces
show_colnames = TRUE, # Afficher nom REL
main = paste("Heatmap transposée (z-score) -", 5, "groupes")
)heatmap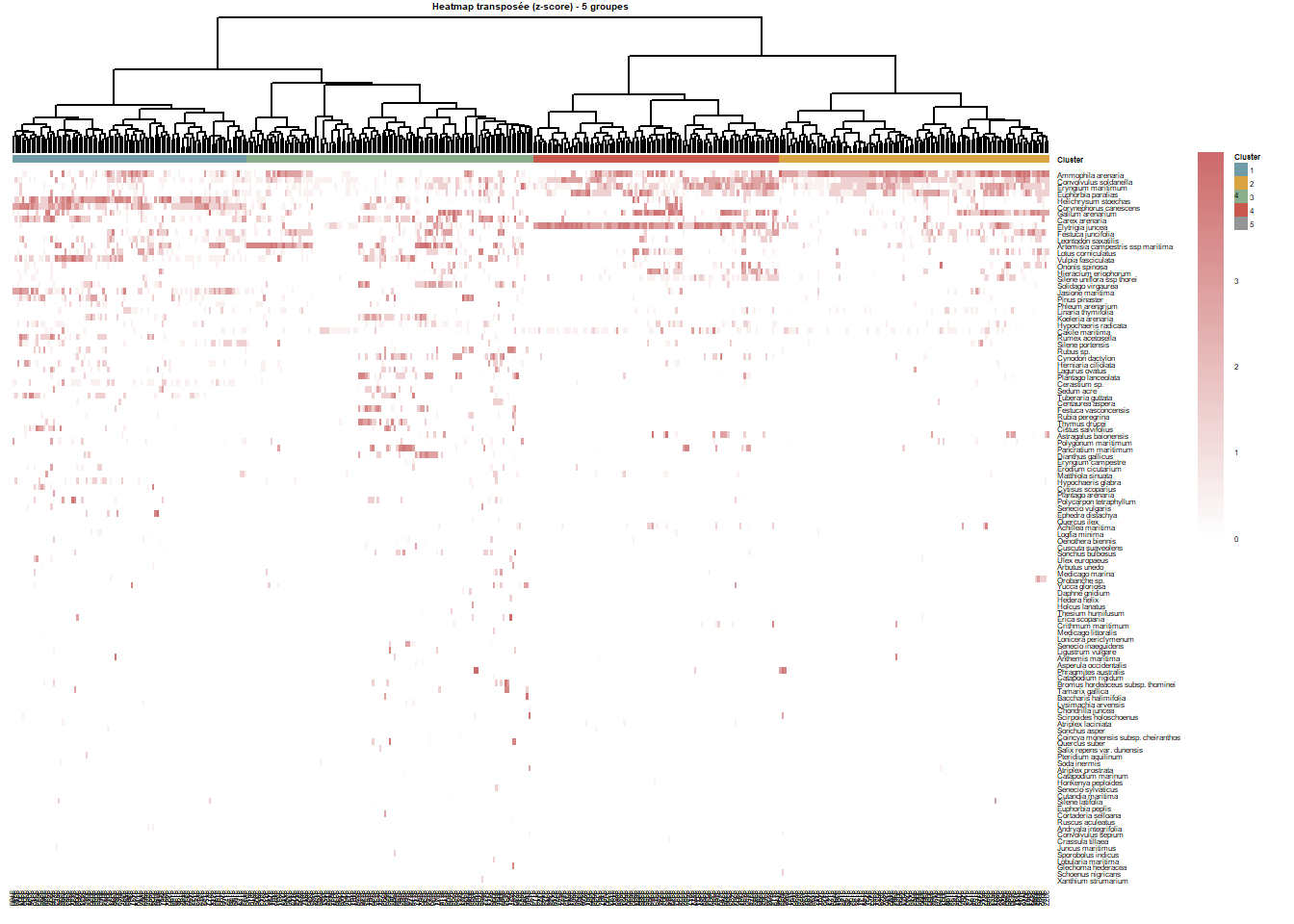
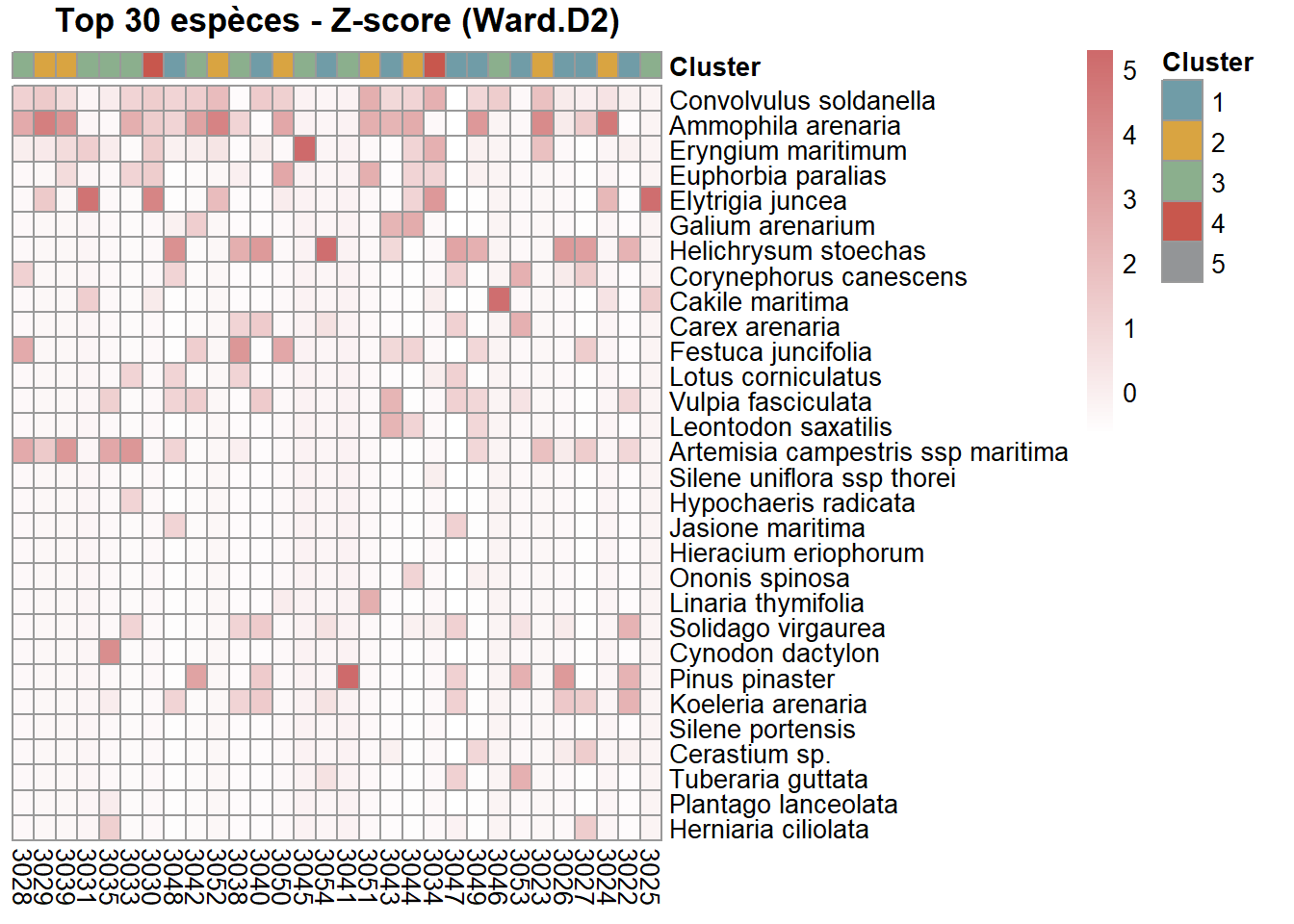
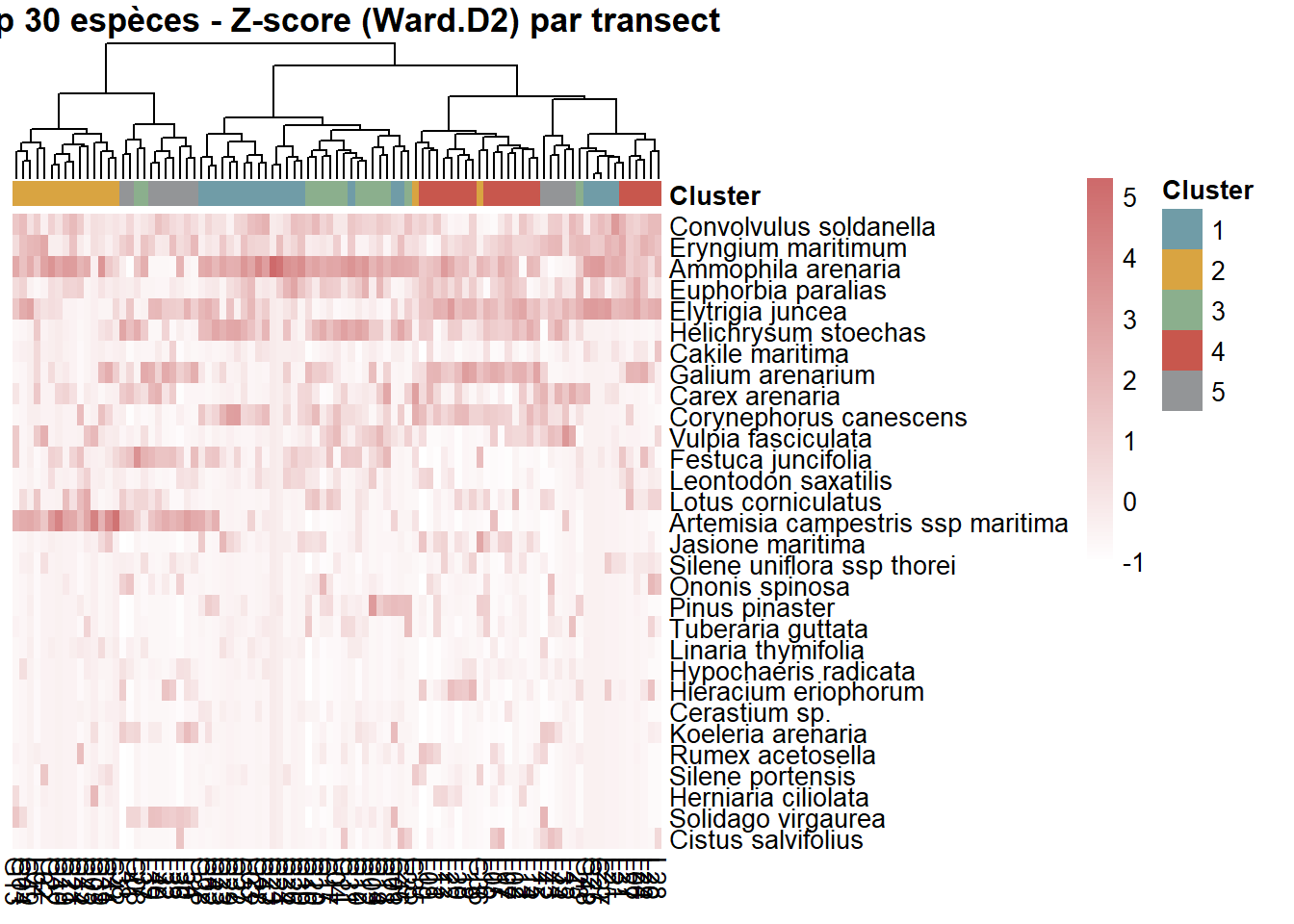
La heatmap est illisble à cause du nombre d’espèces et du nombre du relevés. La heatmap suivante se concentre sur les 30 espèces les plus abondantes.
#### Préparation data ----
top_species <- names(sort(freq, decreasing = TRUE))[1:30]
comm_top <- as.matrix(FLORE_cover_esp_log[, top_species])
comm_top_scaled <- t(scale(t(comm_top)))
comm_top_clean <- comm_top_scaled[complete.cases(comm_top_scaled), ]
comm_top_clean <- comm_top_clean[, complete.cases(t(comm_top_clean))]
rel_clean <- rownames(comm_top_clean)
annotation_clean <- annotation_col[
rownames(annotation_col) %in% rel_clean,
,
drop = FALSE
]
clust_clean <- hclust(dist(t(comm_top_clean)), method = "ward.D2")
ord_cols <- clust_clean$order
#### Visualisation ----
pheatmap(
t(comm_top_clean)[, ord_cols],
color = cols_heat,
cluster_rows = FALSE,
cluster_cols = FALSE,
annotation_col = annotation_clean[ord_cols, , drop = FALSE],
annotation_colors = ann_colors,
show_rownames = TRUE,
show_colnames = TRUE,
main = "Top 30 espèces - Z-score (Ward.D2)"
)
FLORE_cover_esp_transect <- FLORE_cover_esp
FLORE_cover_min <- FLORE_cover[
rowSums(FLORE_cover[, -c(1, 2)], na.rm = TRUE) > 0,
]
FLORE_cover_esp_transect$transect <- FLORE_cover_min$transect_numb
# FLORE_cover_esp_transect <- FLORE_cover_esp_transect[, c("transect", names(FLORE_cover_esp))]
FLORE_transect_esp <- FLORE_cover_esp_transect %>%
group_by(transect) %>%
summarise(across(where(is.numeric), mean, na.rm = TRUE)) %>% # ici, mean car le nombre de transect est différent par transect
ungroup()
# matrice avec nom des lignes = transect
FLORE_transect_esp_mat <- as.matrix(FLORE_transect_esp[, -1])
rownames(FLORE_transect_esp_mat) <- FLORE_transect_esp$transect
# transformation en log
FLORE_transect_log <- log1p(FLORE_transect_esp_mat)
FLORE_transect_log <- FLORE_transect_log[
rowSums(FLORE_transect_log, na.rm = TRUE) > 0,
]#### Préparation data ----
dist_transect <- vegdist(FLORE_transect_log, method = "bray")
clust_transect <- hclust(dist_transect, method = "ward.D2")
groups_transect <- cutree(clust_transect, k = 5)anosim_transect <- anosim(
FLORE_transect_log,
groups_transect,
permutations = 999
)
Call:
anosim(x = FLORE_transect_log, grouping = groups_transect, permutations = 999)
Dissimilarity: bray
ANOSIM statistic R: 0.5902
Significance: 0.001
Permutation: free
Number of permutations: 999simper_transect <- simper(
FLORE_transect_esp_mat,
factor(groups_transect),
permutations = 99
)
characteristic_species_transect <- top5_all(simper_transect, top_n = 5)$`1`
x
Ammophila arenaria Elytrigia juncea
4 4
Helichrysum stoechas Artemisia campestris ssp maritima
3 2
Euphorbia paralias Galium arenarium
2 2
Carex arenaria Eryngium maritimum
1 1
Festuca juncifolia
1
$`2`
x
Ammophila arenaria Artemisia campestris ssp maritima
4 4
Elytrigia juncea Eryngium maritimum
4 2
Galium arenarium Helichrysum stoechas
2 2
Carex arenaria Festuca juncifolia
1 1
$`3`
x
Ammophila arenaria Helichrysum stoechas
4 4
Elytrigia juncea Artemisia campestris ssp maritima
3 2
Euphorbia paralias Festuca juncifolia
2 2
Galium arenarium Carex arenaria
2 1
$`4`
x
Ammophila arenaria Elytrigia juncea
4 4
Galium arenarium Artemisia campestris ssp maritima
4 2
Euphorbia paralias Helichrysum stoechas
2 2
Carex arenaria Eryngium maritimum
1 1
$`5`
x
Ammophila arenaria Artemisia campestris ssp maritima
4 4
Carex arenaria Galium arenarium
4 4
Elytrigia juncea Helichrysum stoechas
3 1 Le diagramme de Venn permet de visualiser les résultats du SIMPER.
Package utilisé
- VennDiagram
#### Préparation data ----
extract_species_filtered_transect <- function(
simper_obj,
top_n = 5,
min_freq = 2
) {
species_list <- list()
for (comp in names(simper_obj)) {
df <- as.data.frame(simper_obj[[comp]])
df <- df[order(-df$average), ] # tri décroissant
top_species <- head(rownames(df), top_n) # top 5
groups <- strsplit(comp, "_")[[1]]
for (g in groups) {
species_list[[g]] <- c(species_list[[g]], top_species)
}
}
lapply(species_list, function(x) {
freq <- sort(table(x), decreasing = TRUE)
names(freq[freq >= min_freq])
})
}
filtered_species_transect <- extract_species_filtered_transect(
simper_transect,
top_n = 5,
min_freq = 2
)
#### Visualisation ----
venn.plot_transect <- venn.diagram(
x = list(
Groupe1 = filtered_species_transect[["1"]],
Groupe2 = filtered_species_transect[["2"]],
Groupe3 = filtered_species_transect[["3"]],
Groupe4 = filtered_species_transect[["4"]],
Groupe5 = filtered_species_transect[["5"]]
),
filename = NULL,
fill = group_palette[as.character(1:5)],
alpha = 0.4,
cex = 1.2,
cat.cex = 0.8,
cat.fontface = "bold",
margin = 0.1
)#### Préparation data ----
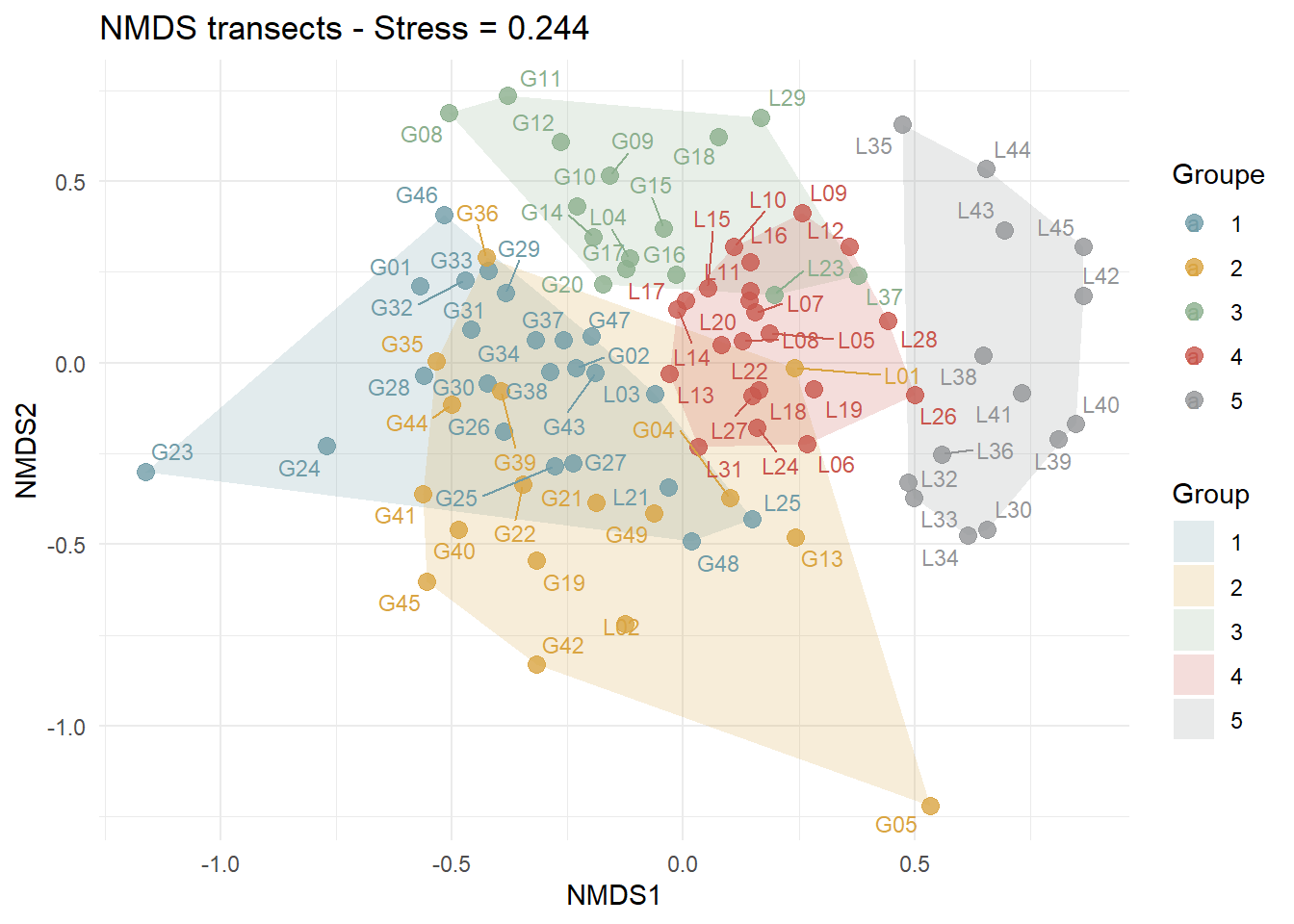
nmds_transect <- metaMDS(
FLORE_transect_log,
distance = "bray",
k = 2,
try = 100
)[1] "Stress = 0.244"nmds_tr_pts <- as.data.frame(scores(nmds_transect, display = "sites"))
nmds_tr_pts$Transect <- rownames(nmds_tr_pts)
nmds_tr_pts$Group <- factor(groups_transect[nmds_tr_pts$Transect])
# Hulls
hulls_tr <- nmds_tr_pts %>%
group_by(Group) %>%
slice(chull(NMDS1, NMDS2))# Fréquences globales
freq_tr <- colSums(FLORE_transect_log > 0, na.rm = TRUE)
top_species_tr <- names(sort(freq_tr, decreasing = TRUE))[1:30]
comm_tr_top <- as.matrix(FLORE_transect_log[, top_species_tr])
comm_tr_scaled <- t(scale(t(comm_tr_top)))
comm_tr_clean <- comm_tr_scaled[complete.cases(comm_tr_scaled), ]
comm_tr_clean <- comm_tr_clean[, complete.cases(t(comm_tr_clean))]
# Annotation transects
annotation_tr <- data.frame(
Cluster = factor(groups_transect[rownames(comm_tr_clean)])
)
rownames(annotation_tr) <- rownames(comm_tr_clean)
# Clustering Ward sur transects (z-scores)
clust_tr_clean <- hclust(dist(comm_tr_clean), method = "ward.D2")
library(pheatmap)
cols_heat <- colorRampPalette(c("#FFFFFF", "#CE6A6B"))(100)
ann_colors_tr <- list(Cluster = group_palette)Package utilisé
- dplyr
- vegan
- ggplot2
La richesse spécifique a été calculée pour chaque relevé puis les résultats ont été emboités à la matrice METADATA.
Richesse spécifique
# Richesse spé par relevé
richesse_rel <- data.frame(
REL = FLORE_cover$REL,
Richesse = rowSums(FLORE_cover[, species_cols] > 0, na.rm = TRUE)
) %>%
mutate(Richesse = Richesse)
METADATA <- METADATA %>%
left_join(richesse_rel, by = "REL")En utilisant le recouvrement moyen (%), l’indice de Shannon a été calculé pour chaque relevé puis les résultats ont été emboités à la matrice METADATA.
Shannon
shannon_rel <- diversity(FLORE_cover[, species_cols], index = "shannon")
METADATA <- METADATA %>%
left_join(
data.frame(REL = FLORE_cover$REL, Shannon = shannon_rel),
by = "REL"
)L’indice de Shannon a ensuite été moyenné par transect.
Shannon par transect
# Shannon par transect
shannon_transect <- METADATA %>%
group_by(transect_numb) %>%
summarise(
shannon_moy = mean(Shannon),
shannon_sd = sd(Shannon),
n_releves = n(),
.groups = "drop"
)En utilisant le recouvrement moyen (%), l’inverse de Simpson a été calculé pour chaque relevé puis les résultats ont été emboités à la matrice METADATA.
Inverse de Simpson
Simpson_inv_rel <- diversity(FLORE_cover[, species_cols], index = "invsimpson")
METADATA <- METADATA %>%
left_join(
data.frame(REL = FLORE_cover$REL, Simpson_inv_rel = Simpson_inv_rel),
by = "REL"
)OUTPUT
- richesse_rel [453 x 2] : richesse spécifique S (0D) par relevé. - shannon_rel [453 x 2] : indice de Shannon H’ (1D) par relevé. - res_shannon [94 x 4] : indice de Shannon H’ (1D) par transect. - Simpson_inv_rel : inverse de Simpson (2D) par relevé.
Enfin, une droite de régression a été tracée entre les deux indices (Shannon ~ Richesse).
shannon ~ richesse
plot(Shannon ~ Richesse, data = METADATA)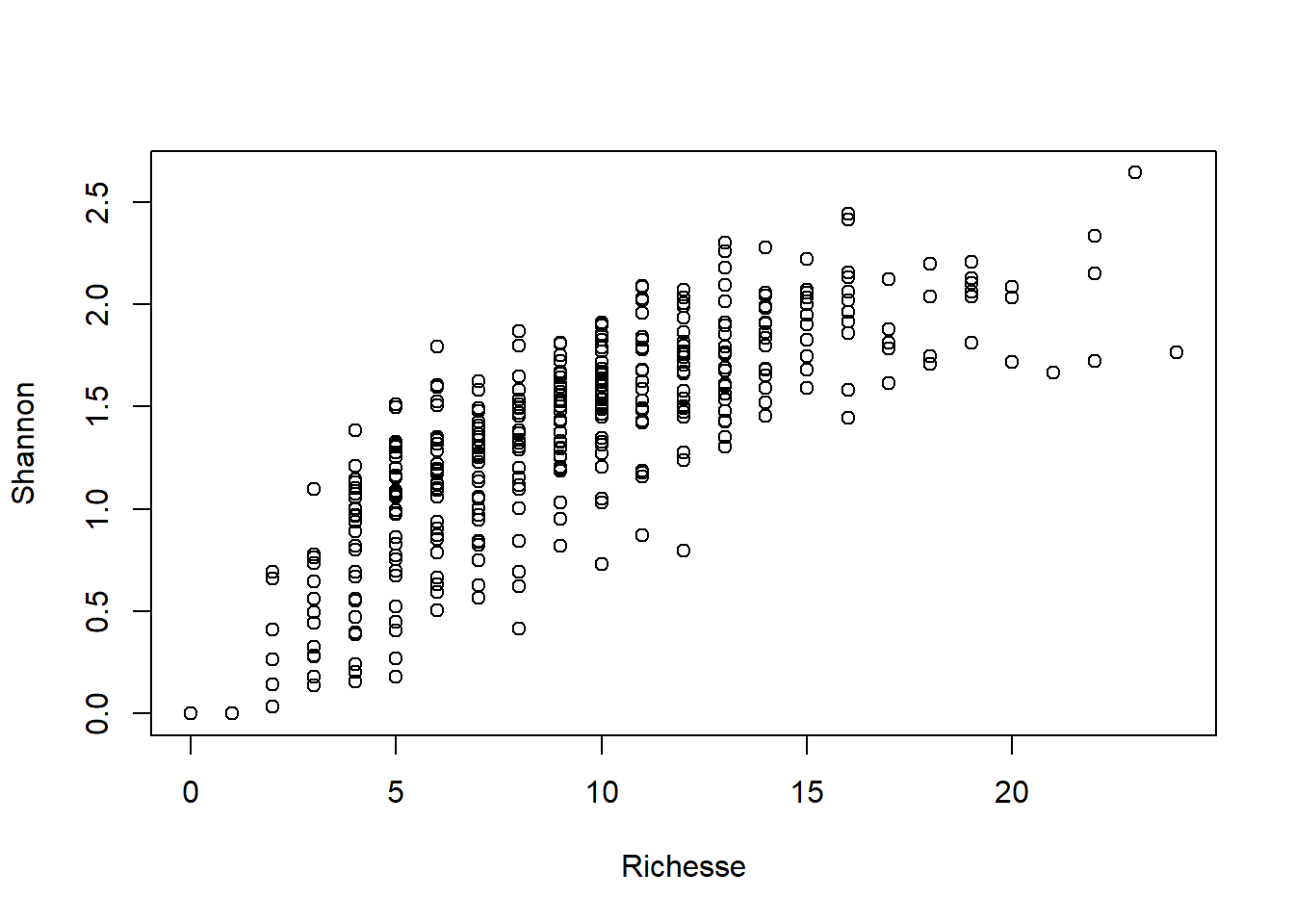
lm(Shannon ~ Richesse, data = METADATA)
Call:
lm(formula = Shannon ~ Richesse, data = METADATA)
Coefficients:
(Intercept) Richesse
0.43714 0.09817 ggplot(METADATA, aes(x = Richesse, y = Shannon)) +
geom_point(alpha = 0.6, color = "bisque3") +
geom_smooth(method = "lm", se = TRUE, color = "aquamarine4", size = 1.2) +
labs(
title = "Richesse spécifique vs Indice de Shannon (2021)",
x = "Richesse spécifique S",
y = "Indice de Shannon H'"
) +
theme_minimal() +
theme(
plot.title = element_text(hjust = 0.5, face = "bold", size = 14)
)Warning: Using `size` aesthetic for lines was deprecated in ggplot2 3.4.0.
ℹ Please use `linewidth` instead.`geom_smooth()` using formula = 'y ~ x'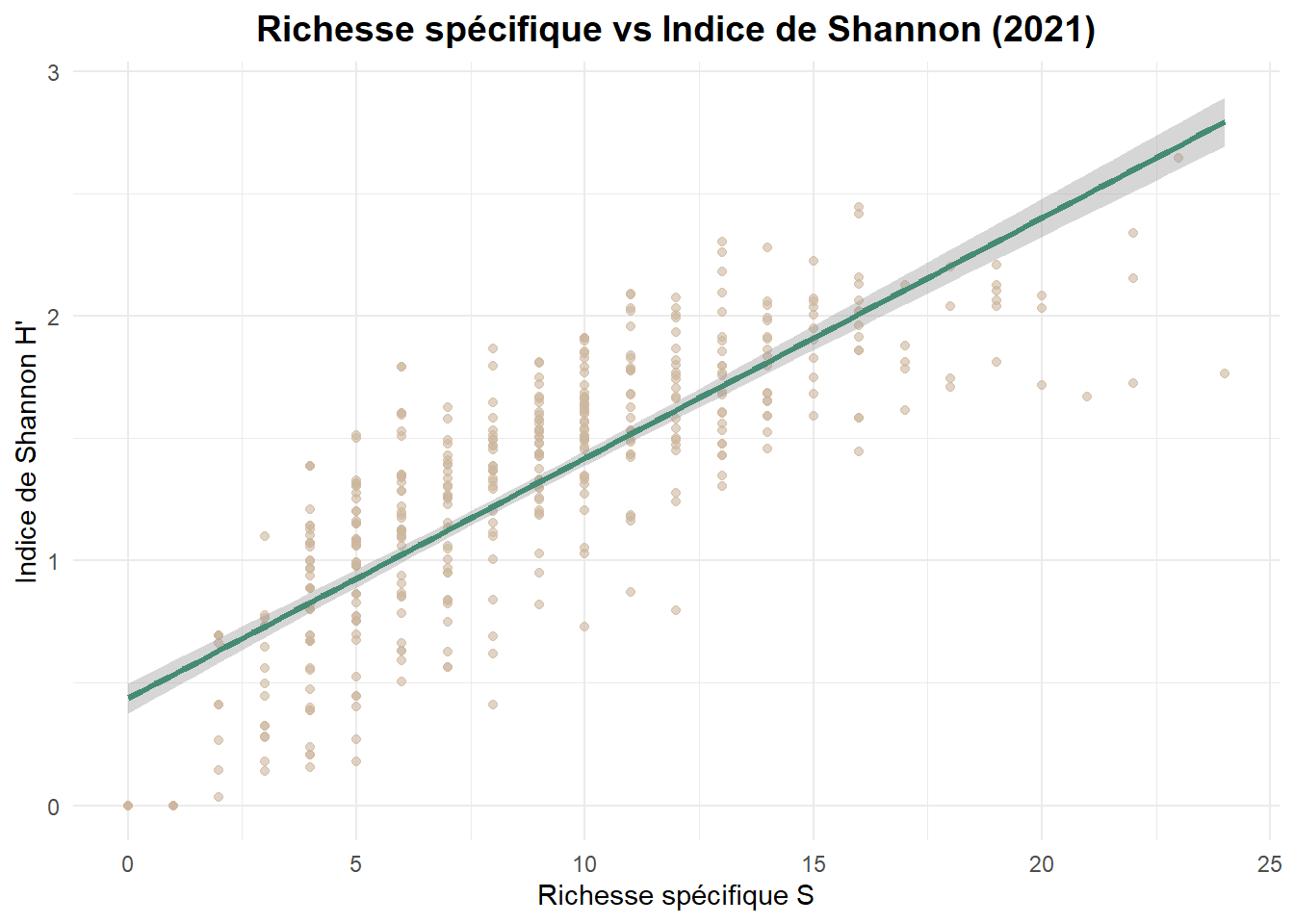
Un tableau est créé pour avoir le nombre de relevés, le nombre d’habitat, le nombre de faciès et la richesse spécifique pour chaque transect.
Package utilisé
- dplyr
# Création d'un dataset
METADATA_transect <- METADATA %>%
group_by(transect_numb) %>%
mutate(
Simpson_inv_rel = ifelse(
is.infinite(Simpson_inv_rel),
Richesse,
Simpson_inv_rel
)
) %>%
summarise(
n_releves = n(),
n_habitats = n_distinct(Type_habitat, na.rm = TRUE),
n_facies = n_distinct(id_facies, na.rm = TRUE),
Richesse_moy = mean(Richesse, na.rm = TRUE),
Richesse_sd = sd(Richesse, na.rm = TRUE),
Shannon_moy = mean(Shannon, na.rm = TRUE),
Shannon_sd = sd(Shannon, na.rm = TRUE),
Simpson_moy = mean(Simpson_inv_rel, na.rm = TRUE),
Simpson_sd = sd(Simpson_inv_rel, na.rm = TRUE),
.groups = "drop"
) %>%
arrange(transect_numb)OUTPUT
- METADATA_transect [94 x 4] : pour chaque transect, nombre de relevés, nombre d’habitats, nombre de faciès et les indices 0D, 1D et 2D (moyenne et écart-type).
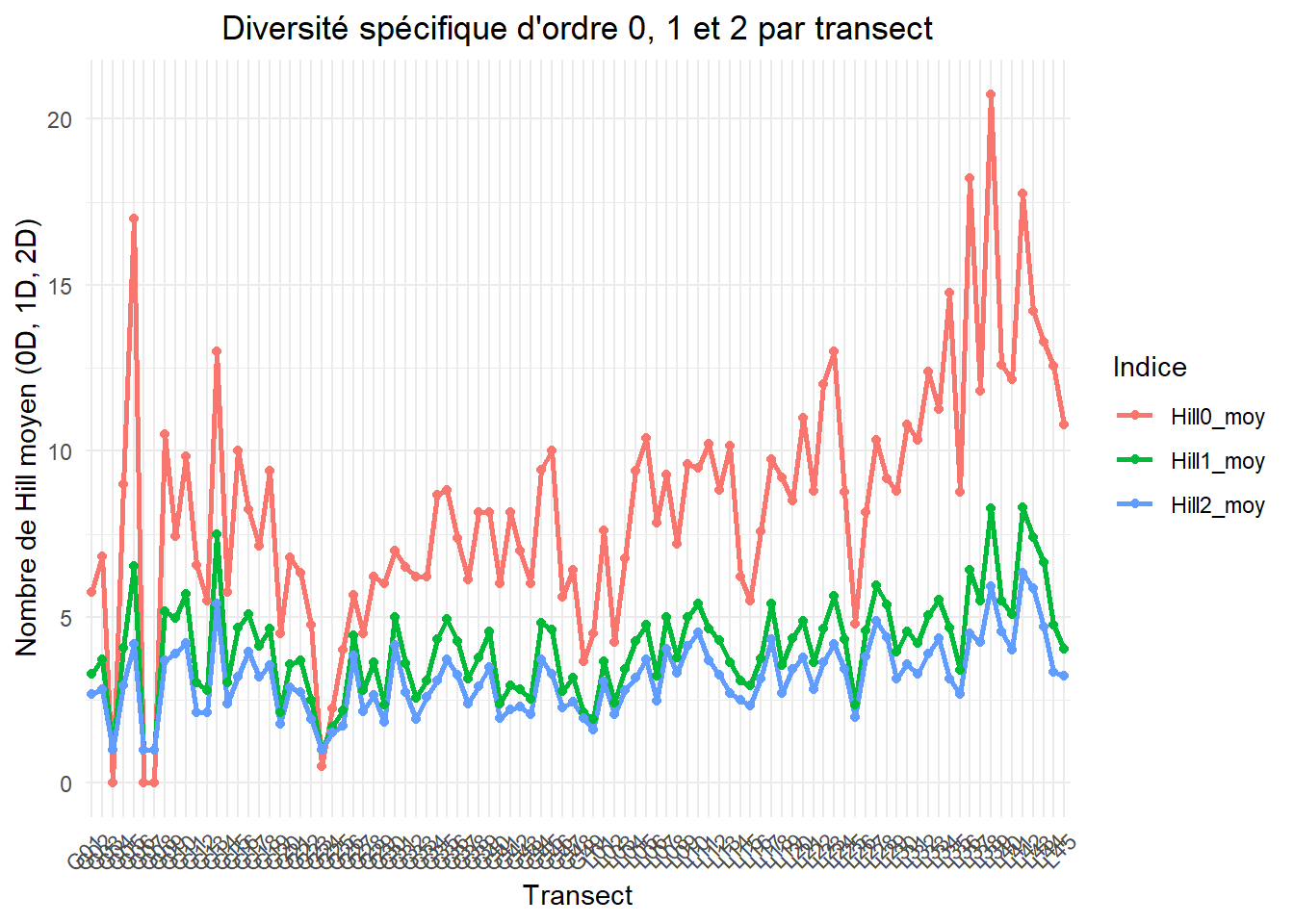
Les nombres de Hill permettent de mesurer la diversité d’une communauté de manière cohérente entre indices et directement interprétable en nombre d’espèces effectives. Ils mettent la richesse spécifique, Shannon et Simpson sur la même échelle (ordres q), comparables entre eux.
Packages utilisés
- dplyr
- tidyr
#### Diversité spécifique ----
Hill_0s <- richesse_rel$Richesse
Hill_1s <- exp(shannon_rel)
Hill_2s <- Simpson_inv_rel
#### Dataset Hill Spécifique
Hill_spe_transect <- METADATA %>%
group_by(transect_numb) %>%
mutate(
Simpson_inv_corr = ifelse(is.infinite(Simpson_inv_rel), 1, Simpson_inv_rel)
) %>%
# Résultats 'inf' dans les valeurs de l'inverse de Simpson donc correction
summarise(
Hill0_moy = mean(Richesse, na.rm = TRUE),
Hill0_sd = sd(Richesse, na.rm = TRUE),
Hill1_moy = mean(exp(Shannon), na.rm = TRUE),
Hill1_sd = sd(exp(Shannon), na.rm = TRUE),
Hill2_moy = mean(Simpson_inv_corr, na.rm = TRUE), # Utilisation de la correction
Hill2_sd = sd(Simpson_inv_corr, na.rm = TRUE), # pour ne plus avoir de 'inf'
n_releves = n(),
.groups = "drop"
)
Hill_transect_long <- Hill_spe_transect %>%
select(transect_numb, Hill0_moy, Hill1_moy, Hill2_moy) %>%
pivot_longer(
cols = starts_with("Hill"),
names_to = "Indice",
values_to = "Valeur"
)OUTPUT
- Hill_xs : vecteurs des 3 ordres de nombres de Hill représentant la richesse spécifique (0D), l’indice de Shannon en exp (1D) et l’inverse de Simpson (2D).
- Hill_transect : dataframe des indices de diversité spéicifique sous nombres de Hill par transect.
- Hill_transect_long : dataframe en long.
Package utilisé
- ggplot2
graph_diversite <- ggplot(
Hill_transect_long,
aes(x = transect_numb, y = Valeur, color = Indice, group = Indice)
) +
geom_line(size = 1) +
geom_point(size = 1.5) +
labs(
x = "Transect",
y = "Nombre de Hill moyen (0D, 1D, 2D)",
title = "Diversité spécifique d'ordre 0, 1 et 2 par transect"
) +
theme_minimal() +
theme(
axis.text.x = element_text(angle = 45, hjust = 1),
plot.title = element_text(hjust = 0.5)
)cor(
Hill_spe_transect$Hill0_moy,
Hill_spe_transect$Hill1_moy,
use = "complete.obs"
)[1] 0.9037351cor(
Hill_spe_transect$Hill0_moy,
Hill_spe_transect$Hill2_moy,
use = "complete.obs"
)[1] 0.8512431cor(
Hill_spe_transect$Hill1_moy,
Hill_spe_transect$Hill2_moy,
use = "complete.obs"
)[1] 0.9834085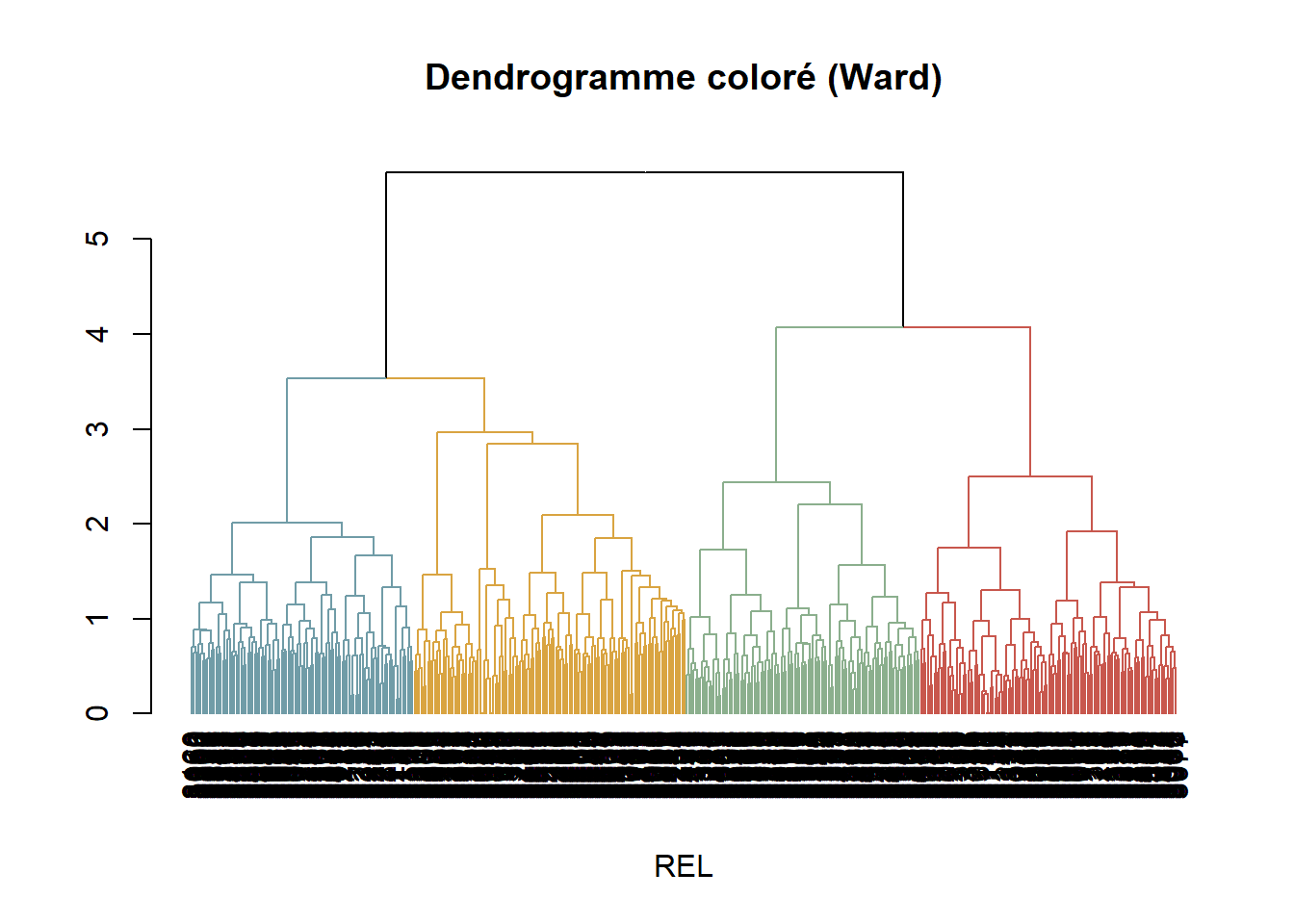
{kind=link}
{kind=link}