Code
source("03.script/00_packages.R")source("03.script/00_packages.R")Importation des données de terrain issues du suivi des communautés dunaires.
Package utilisé
- RColorBrewer
# Palette de couleur
MyCol8 <- brewer.pal(n = 8, name = "Dark2")
# Séparation des chemins "data" (chemin) et "resultats" (path_res)
chemin <- "D:/Master 2/S10/stagem2_lcodoner/02.data/DuneBioDiv_GenetData"
path_res <- "D:/Master 2/S10/stagem2_lcodoner/04.results"
terdat <- read.table(
file.path(chemin, "AllDataTerrain_forR.txt"),
sep = "\t",
header = TRUE
)OUTPUT
- terdat [3274 x 9] : pour chaque individu, description de son site, son faciès et ses coordonnées géographiques
source("03.script/03_functions.R")OUTPUT
- opérateur %!in%
- load_genetics()
- analyse_espece_fst()
- compute_all_hill()
Structuration des données terrain. Répartition des individus par espècs et par site.
table(terdat$species, terdat$site)
G04 G10 G12 G13 G19 G20 G21 G29 G30 G31 G37 G38 G40 G46 G47 G49 L10 L11
AA 16 15 15 15 15 17 15 15 15 17 15 16 14 15 16 16 15 16
AC 15 17 0 15 15 0 15 0 0 0 16 15 15 15 15 15 0 15
CS 15 15 15 0 15 19 15 15 15 16 15 15 15 15 15 15 15 15
CV 14 15 15 16 15 15 15 15 15 15 15 15 15 15 15 15 15 16
EJ 15 0 15 15 15 15 15 15 15 15 15 15 15 16 15 15 16 15
EM 15 15 14 15 16 15 15 16 15 15 15 16 15 15 15 15 15 15
EP 16 15 15 15 15 16 15 17 15 16 15 13 15 15 15 16 15 15
HS 16 15 15 0 19 15 15 18 15 16 15 16 15 16 15 15 15 15
L12 L21 L22 L24 L30 L31 L32 L36 L37 L38 L43
AA 16 17 16 15 15 17 14 15 16 15 8
AC 0 0 0 0 15 0 17 15 16 15 16
CS 16 15 14 0 15 15 0 15 0 0 15
CV 15 15 14 16 17 15 16 15 15 15 15
EJ 15 17 16 15 16 15 15 15 16 15 15
EM 15 15 15 15 15 15 15 15 22 15 15
EP 16 15 17 15 15 15 16 15 16 15 15
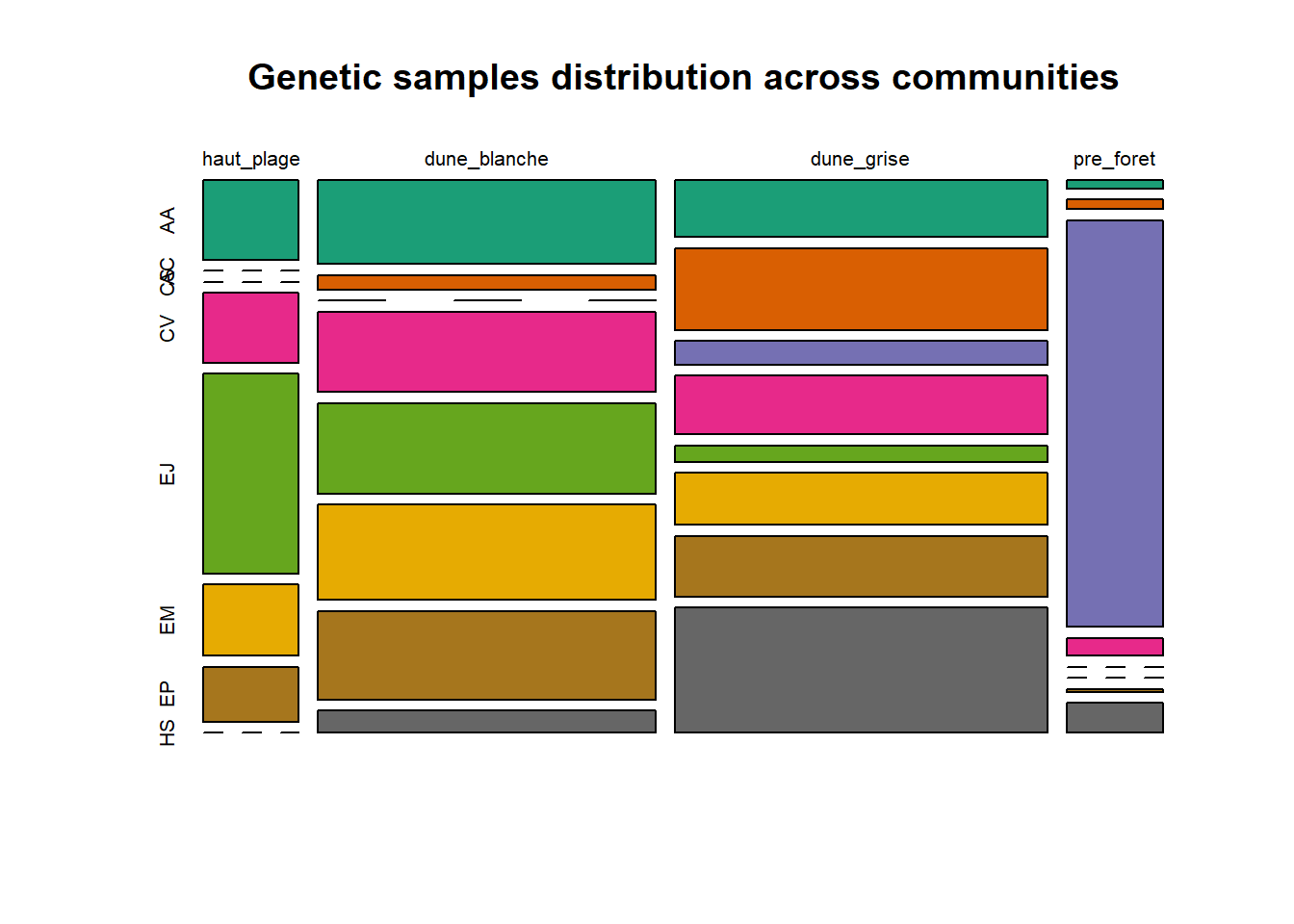
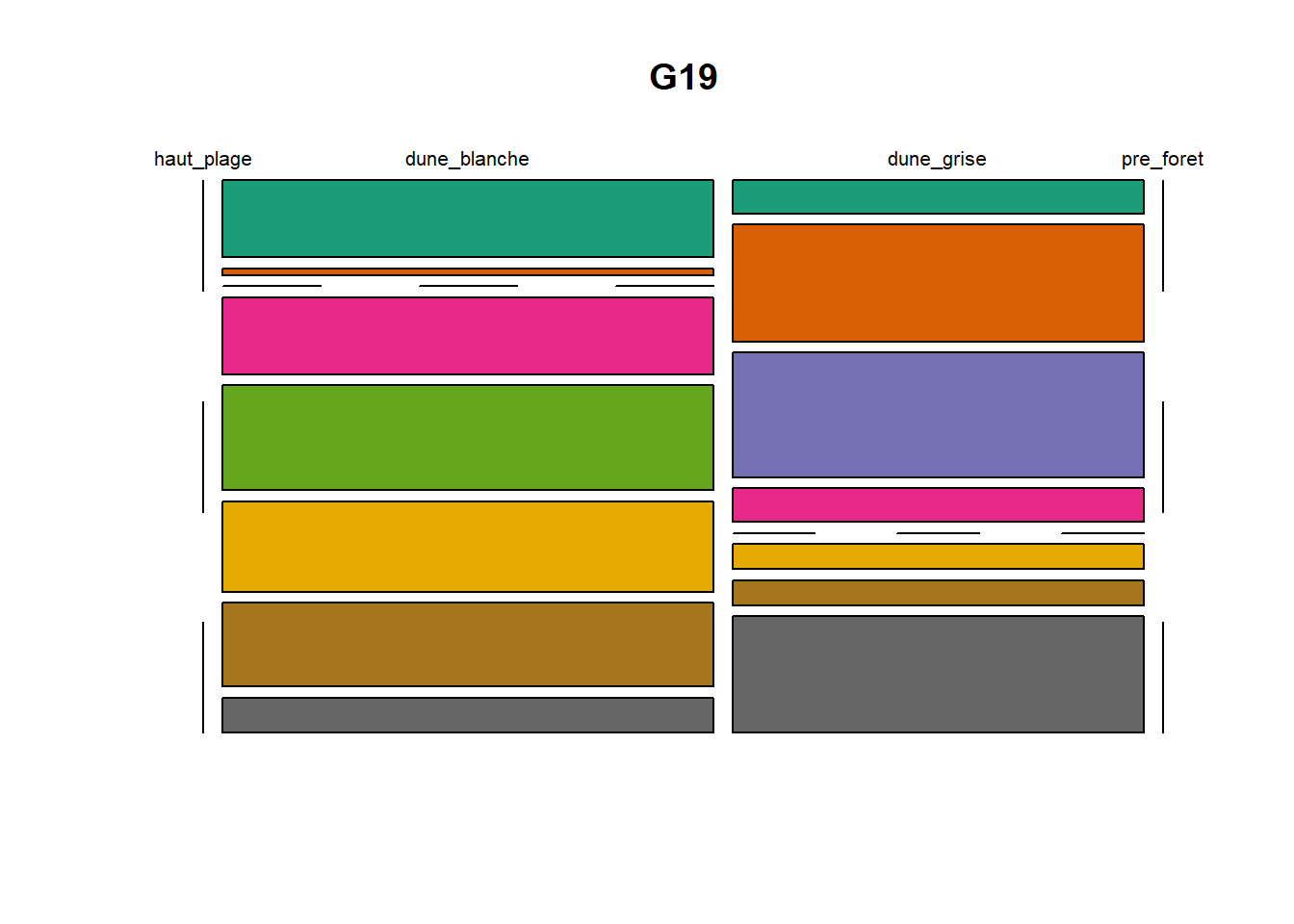
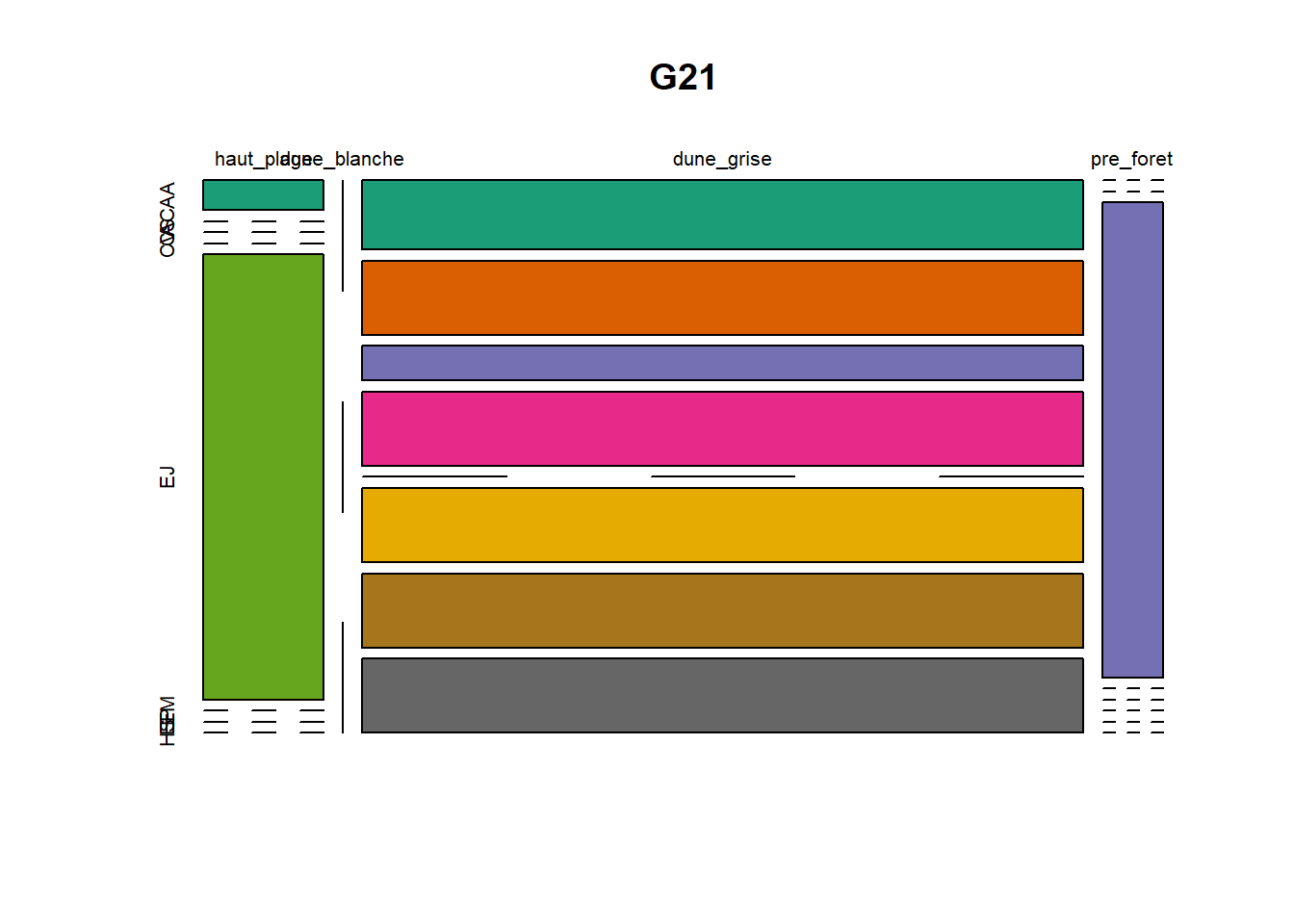
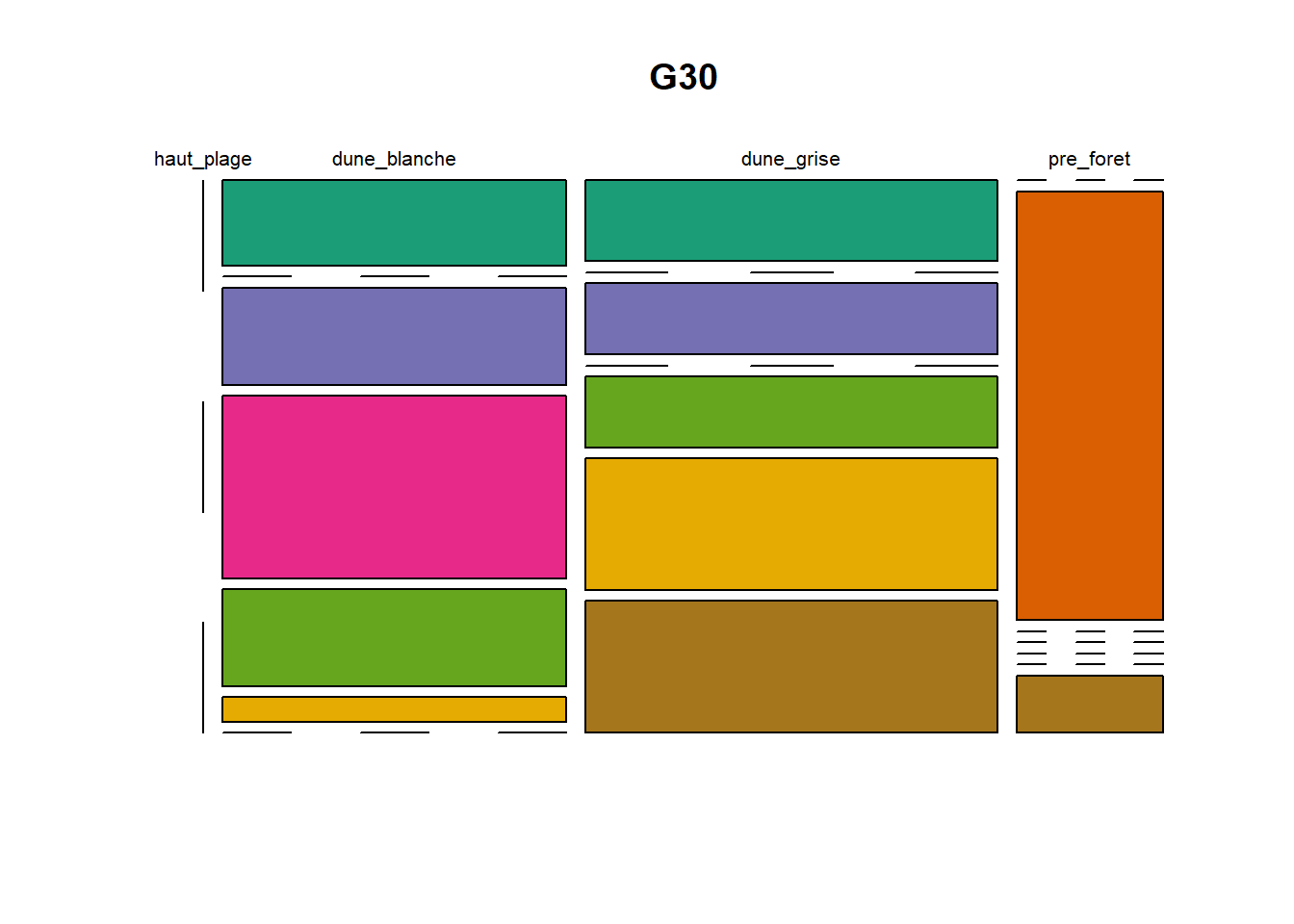
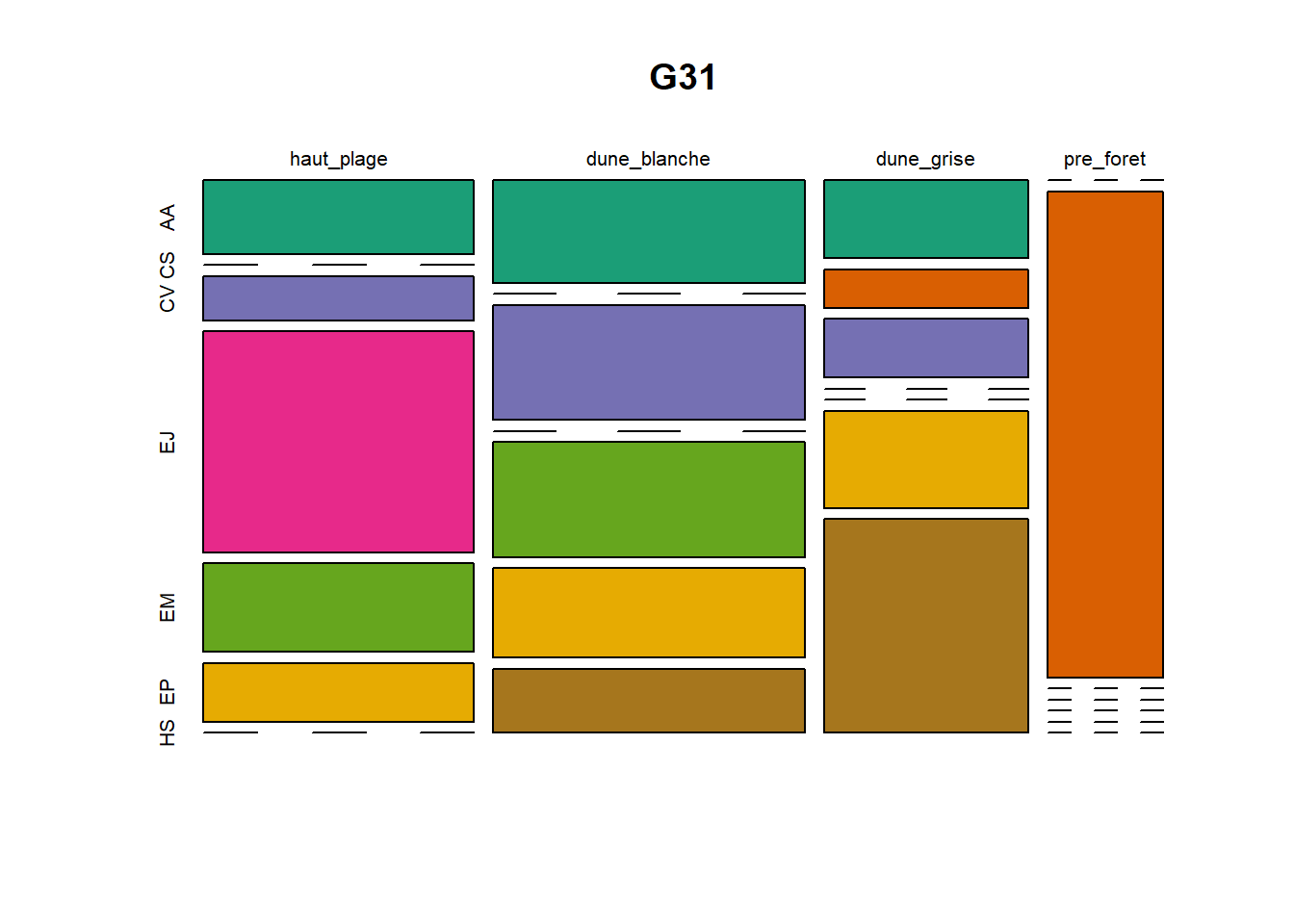
HS 16 15 16 16 15 15 15 15 15 15 17Synthèse graphique du nombre d’individus par espèces et par faciès dunaire (haut de plage, dune blanche, dune grise, pré-forêt).
specom <- table(terdat$species, terdat$community)
specom <- as.table(cbind(
haut_plage = specom[, "haut_plage"],
dune_blanche = specom[, "dune_blanche"],
dune_grise = specom[, "dune_grise"],
pre_foret = specom[, "pre_foret"]
))Ensuite, pour chaque site, construction d’un barplot local espèces x faciès
# graphique par site
specRecAll <- c()
par(mfrow = c(6, 5), mar = c(3, 3, 1, 1) + 0.1)for (loc in unique(terdat$site)) {
spec <- table(
terdat[terdat$site == loc, ]$species,
terdat[terdat$site == loc, ]$community
)
if ("haut_plage" %!in% colnames(spec)) {
spec <- cbind(spec, "haut_plage" = 0)
}
if ("dune_blanche" %!in% colnames(spec)) {
spec <- cbind(spec, "dune_blanche" = 0)
}
if ("dune_grise" %!in% colnames(spec)) {
spec <- cbind(spec, "dune_grise" = 0)
}
if ("pre_foret" %!in% colnames(spec)) {
spec <- cbind(spec, "pre_foret" = 0)
}
spec <- as.table(spec[, c(
"haut_plage",
"dune_blanche",
"dune_grise",
"pre_foret"
)])
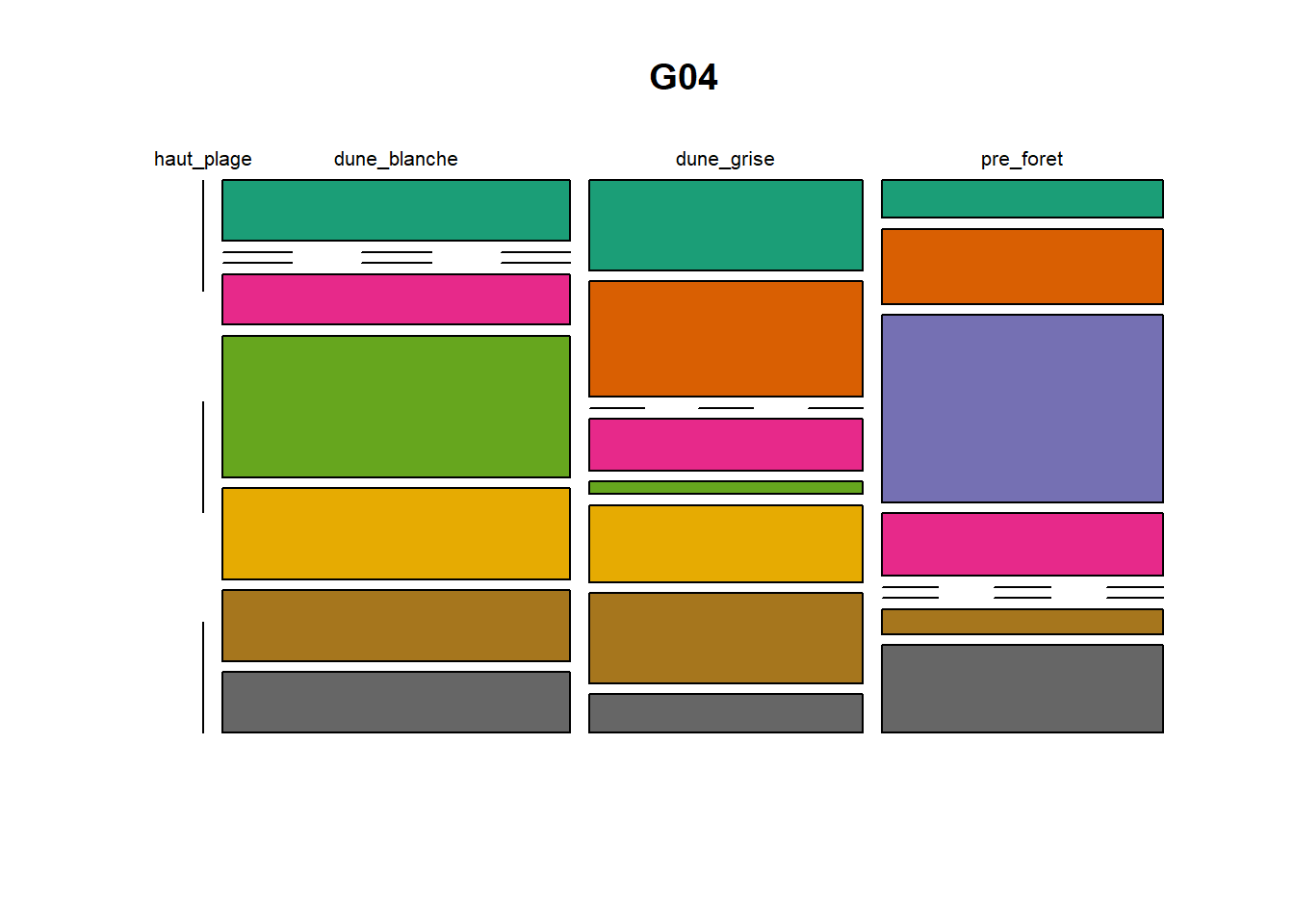
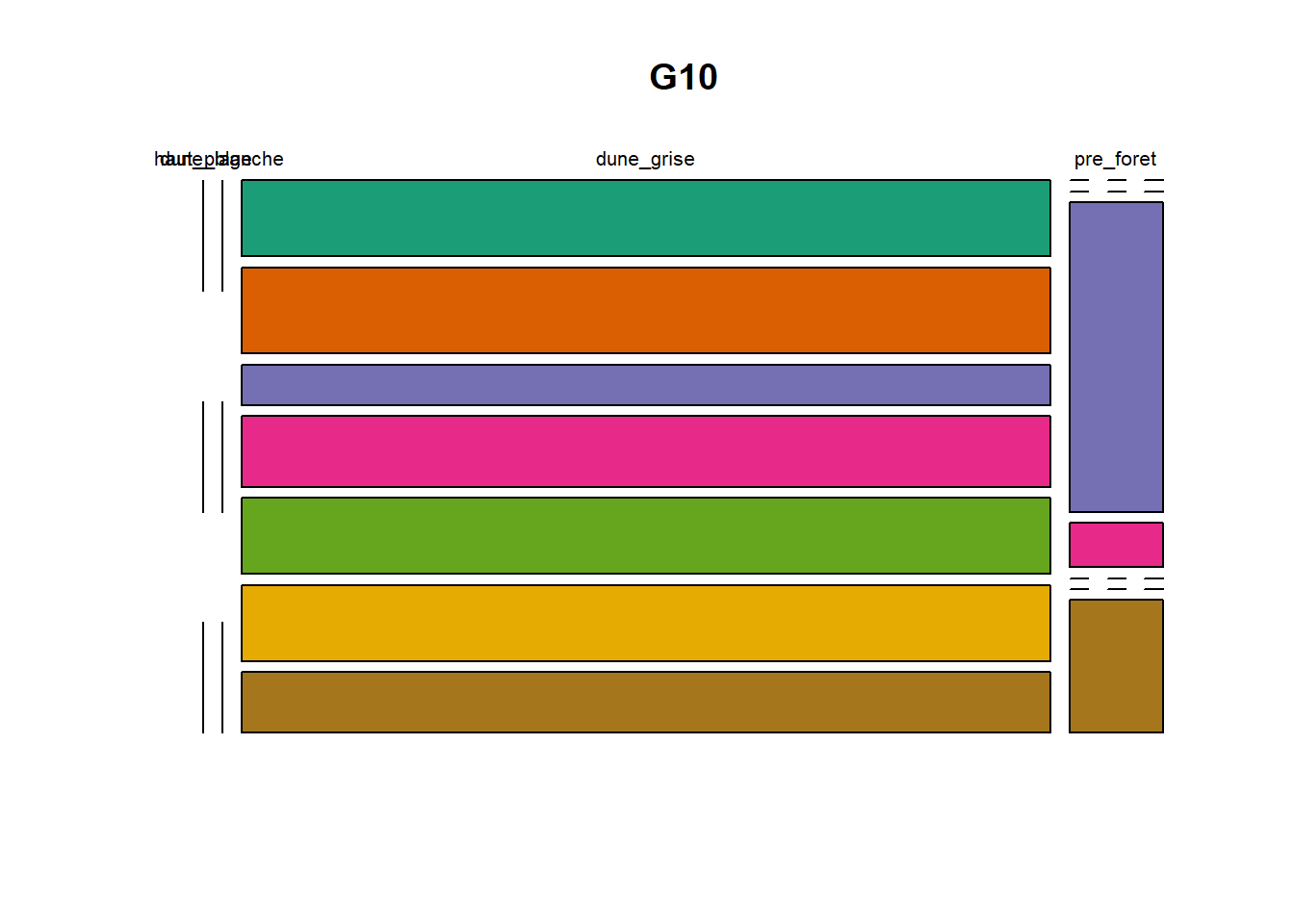
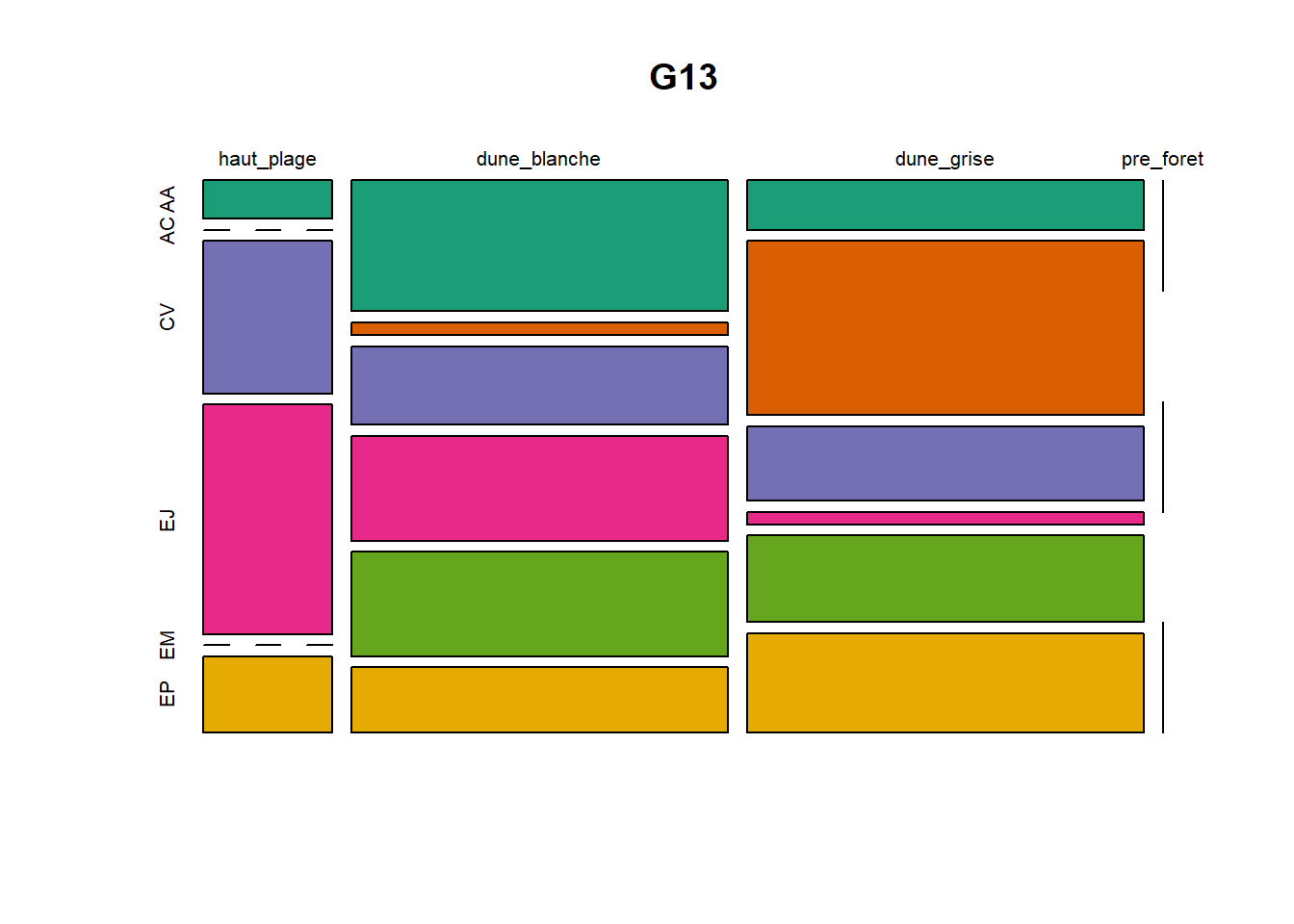
plot(t(spec), col = MyCol8, main = loc)
specRec <- cbind(Site = loc, Species = rownames(spec), spec)
specRecAll <- rbind(specRecAll, specRec)
}




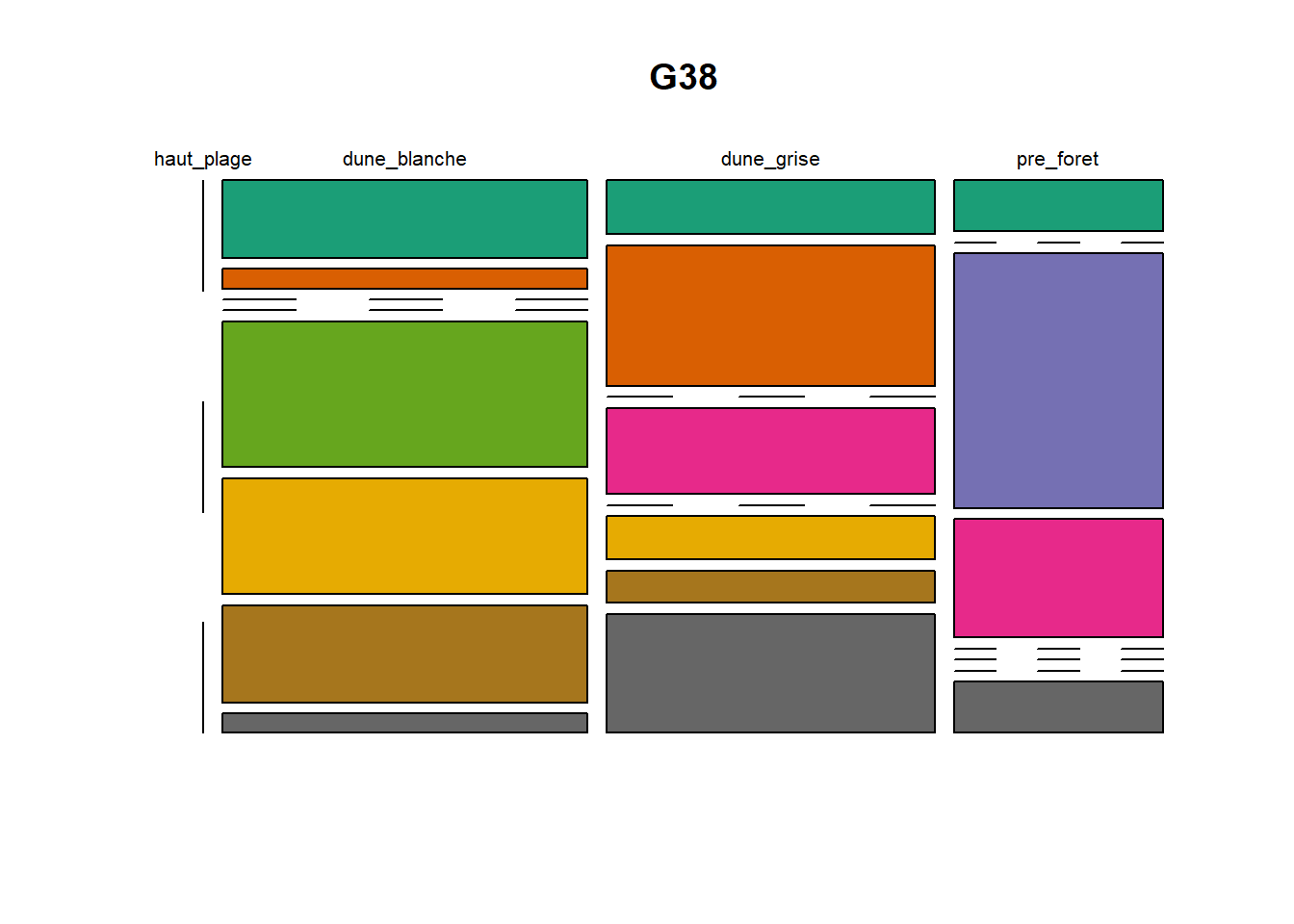

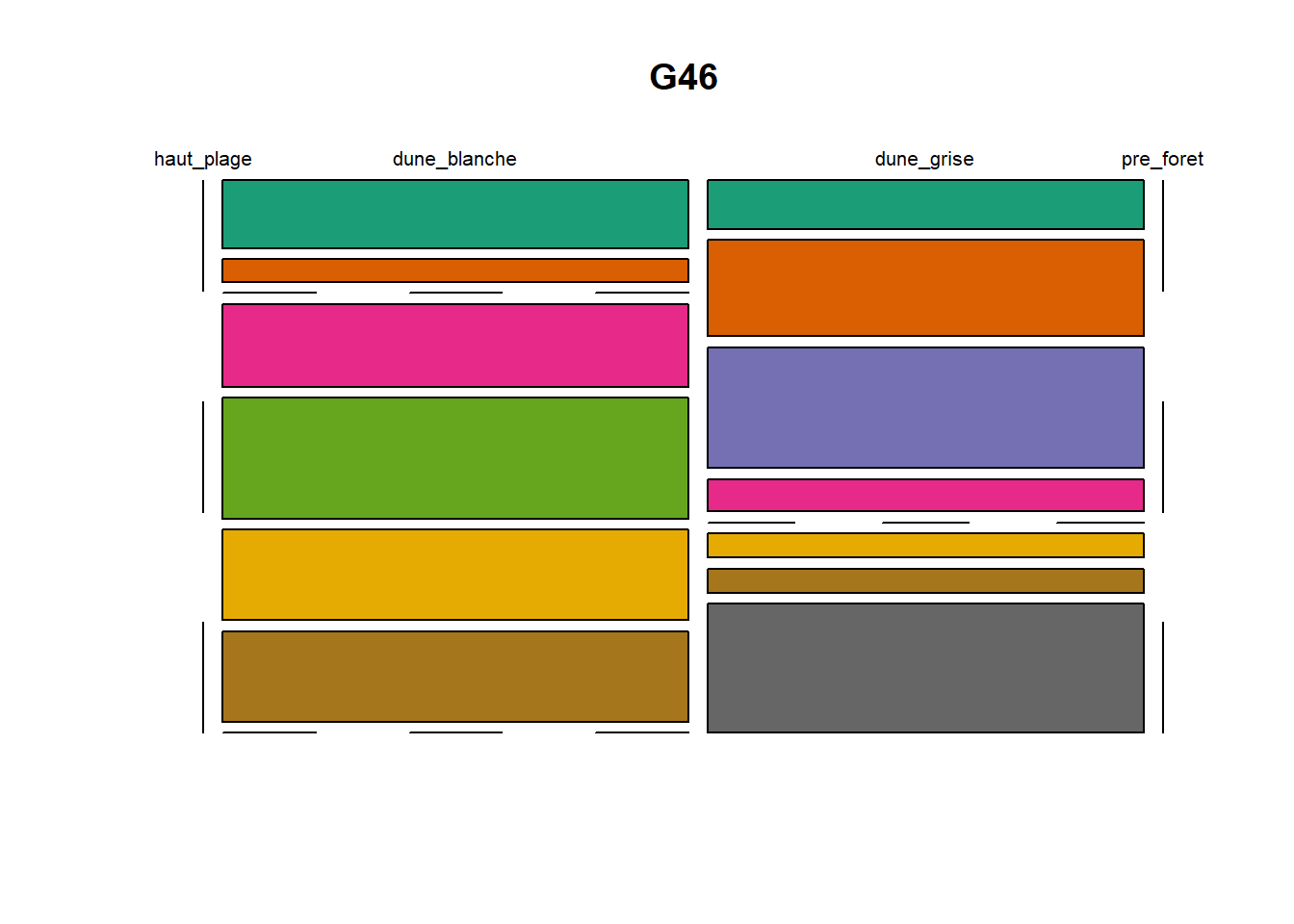
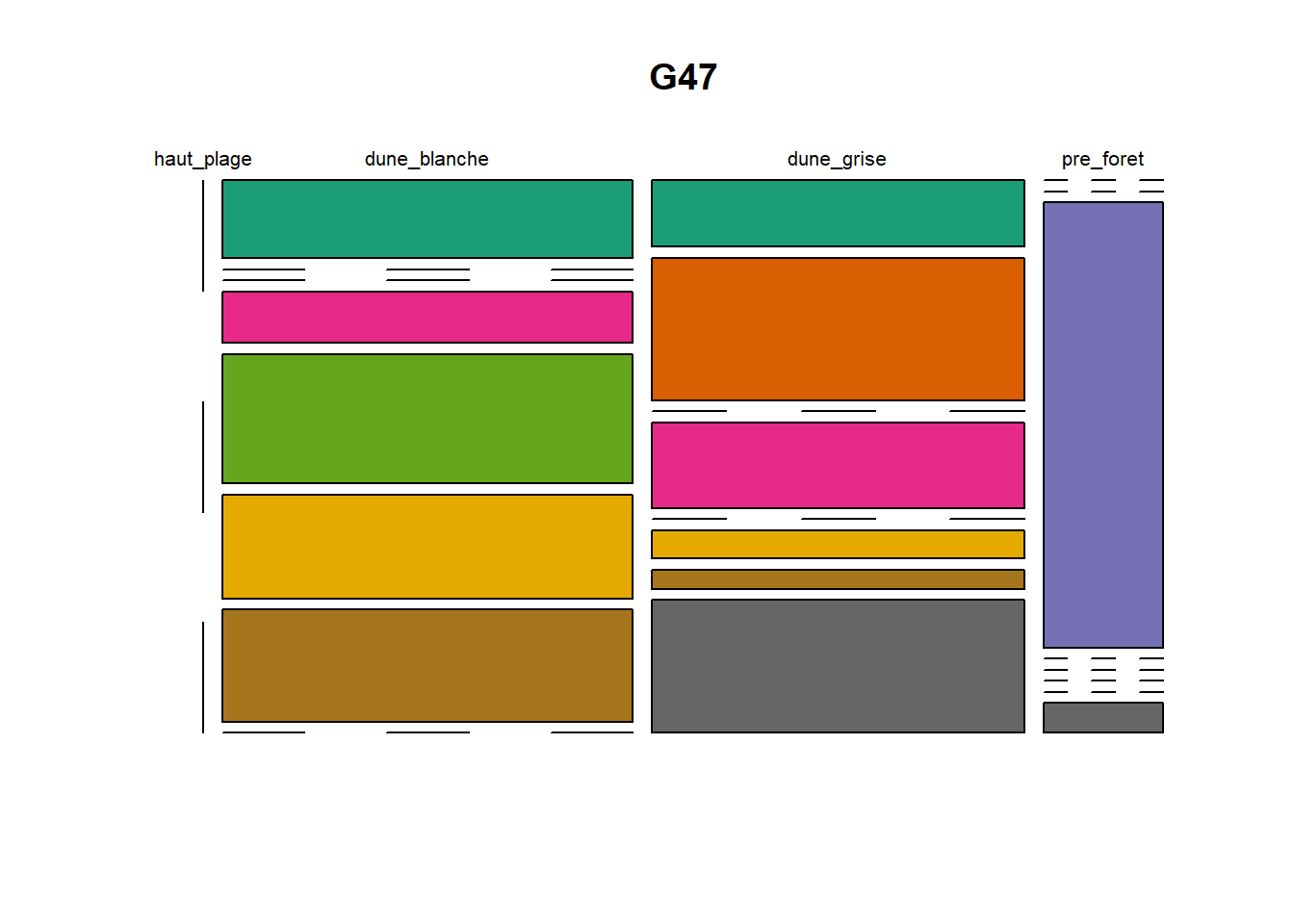

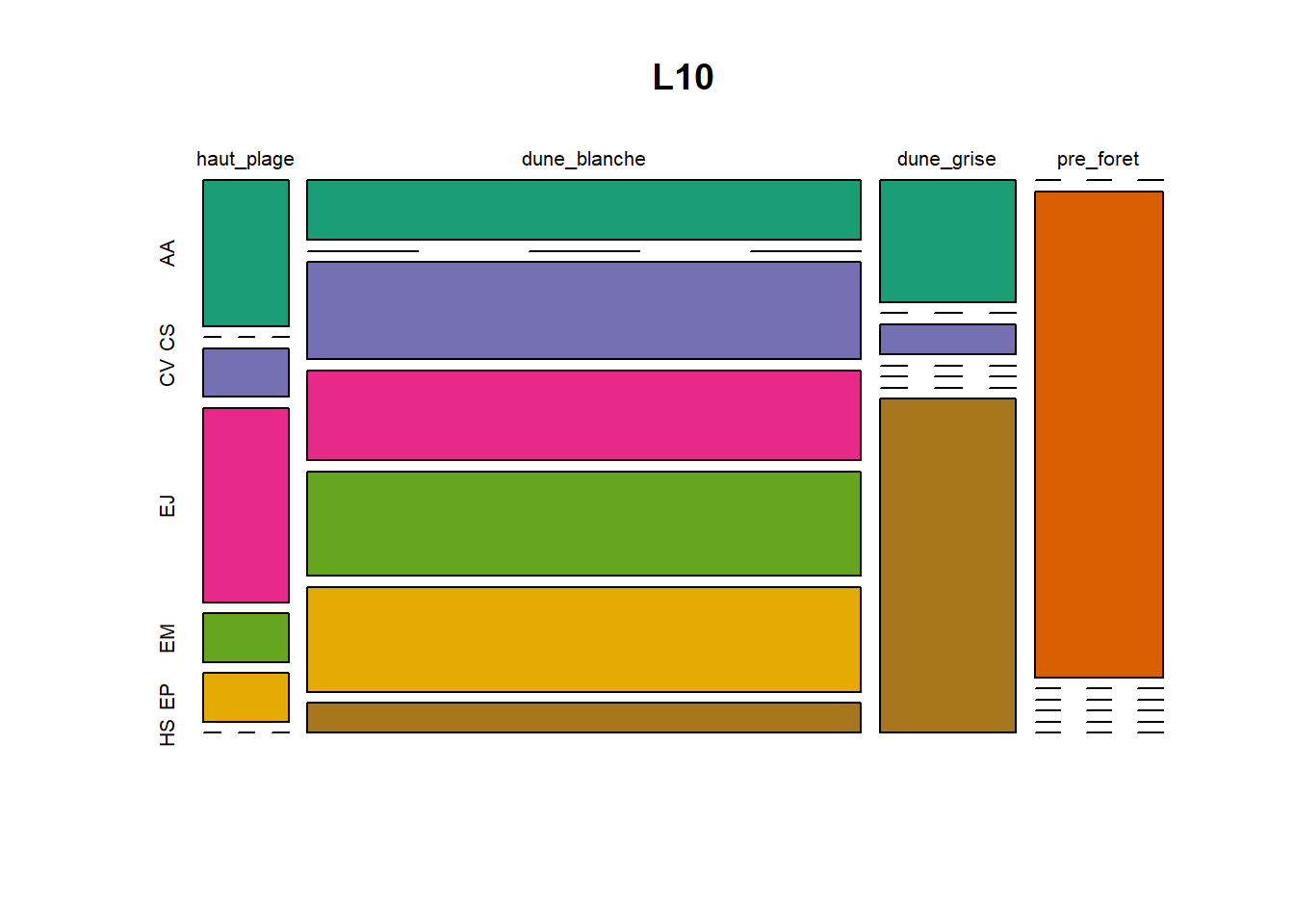
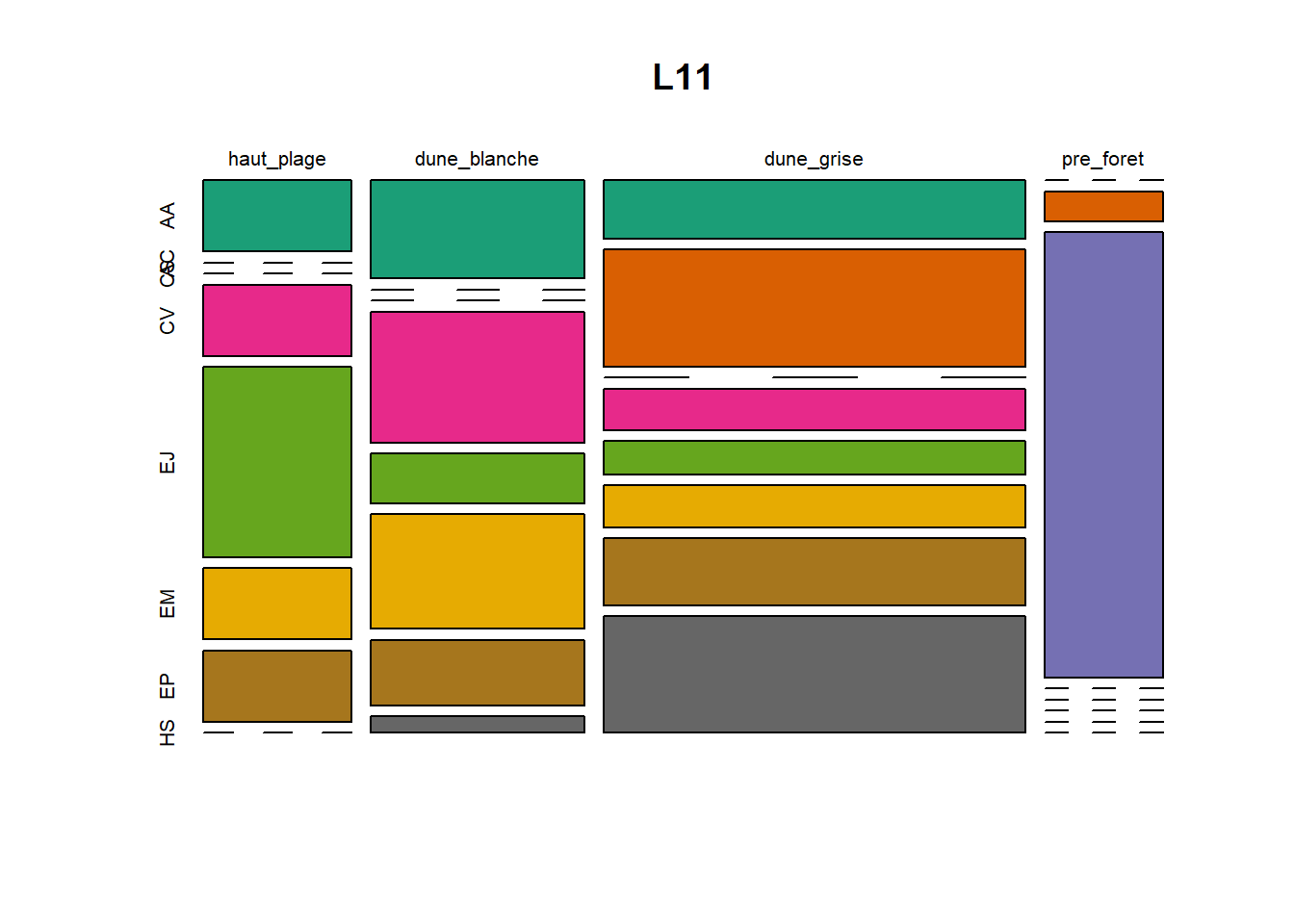
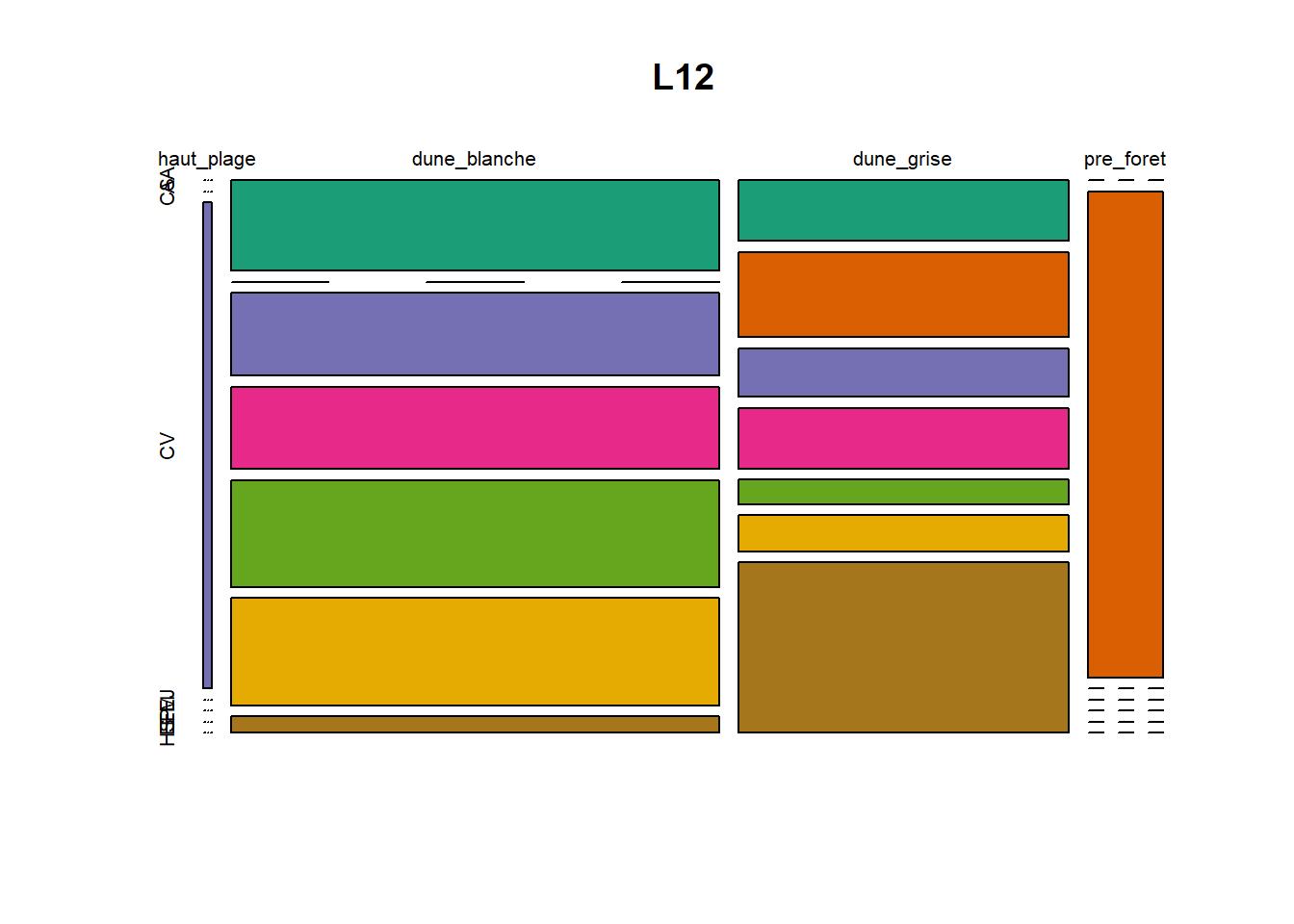


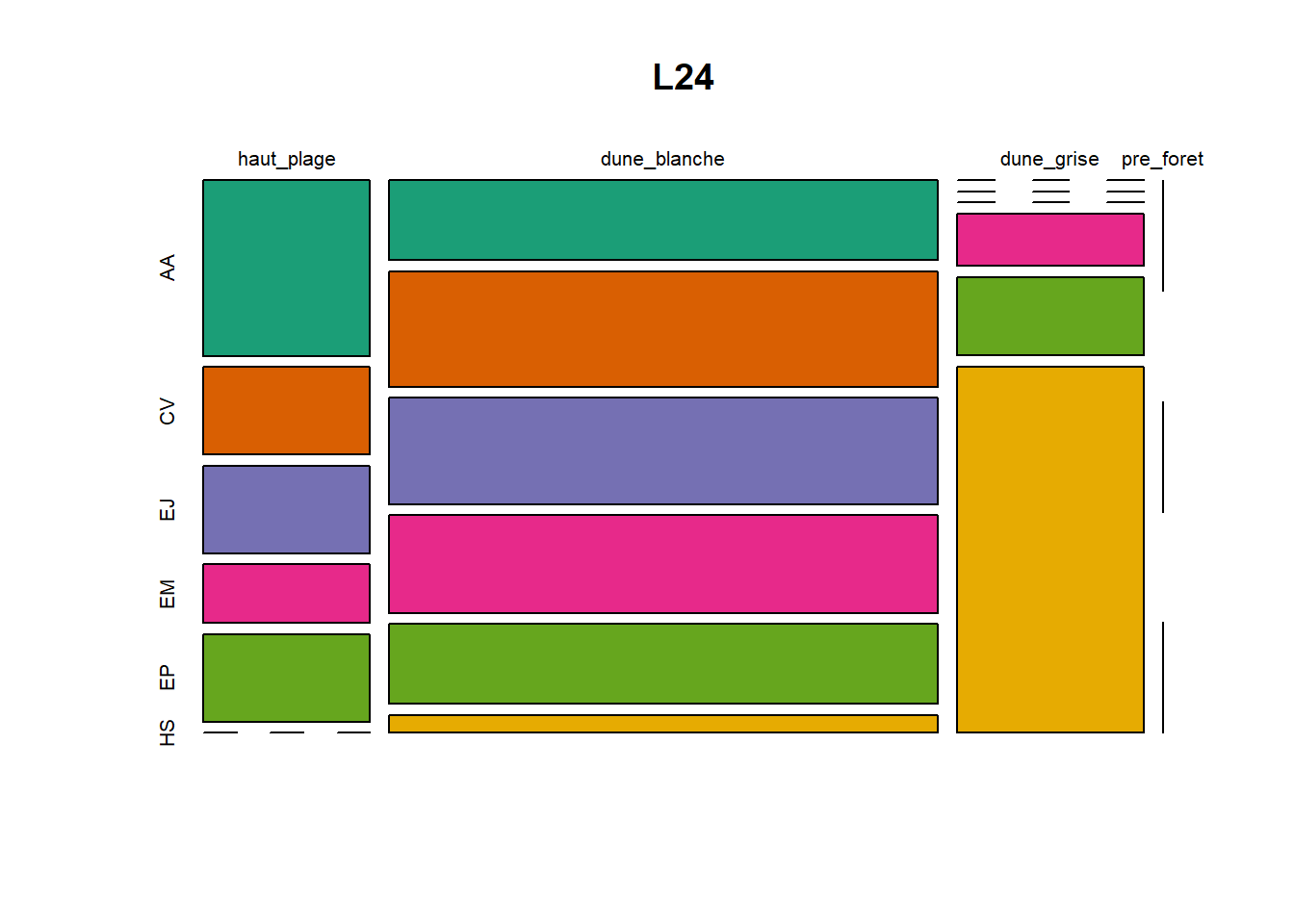

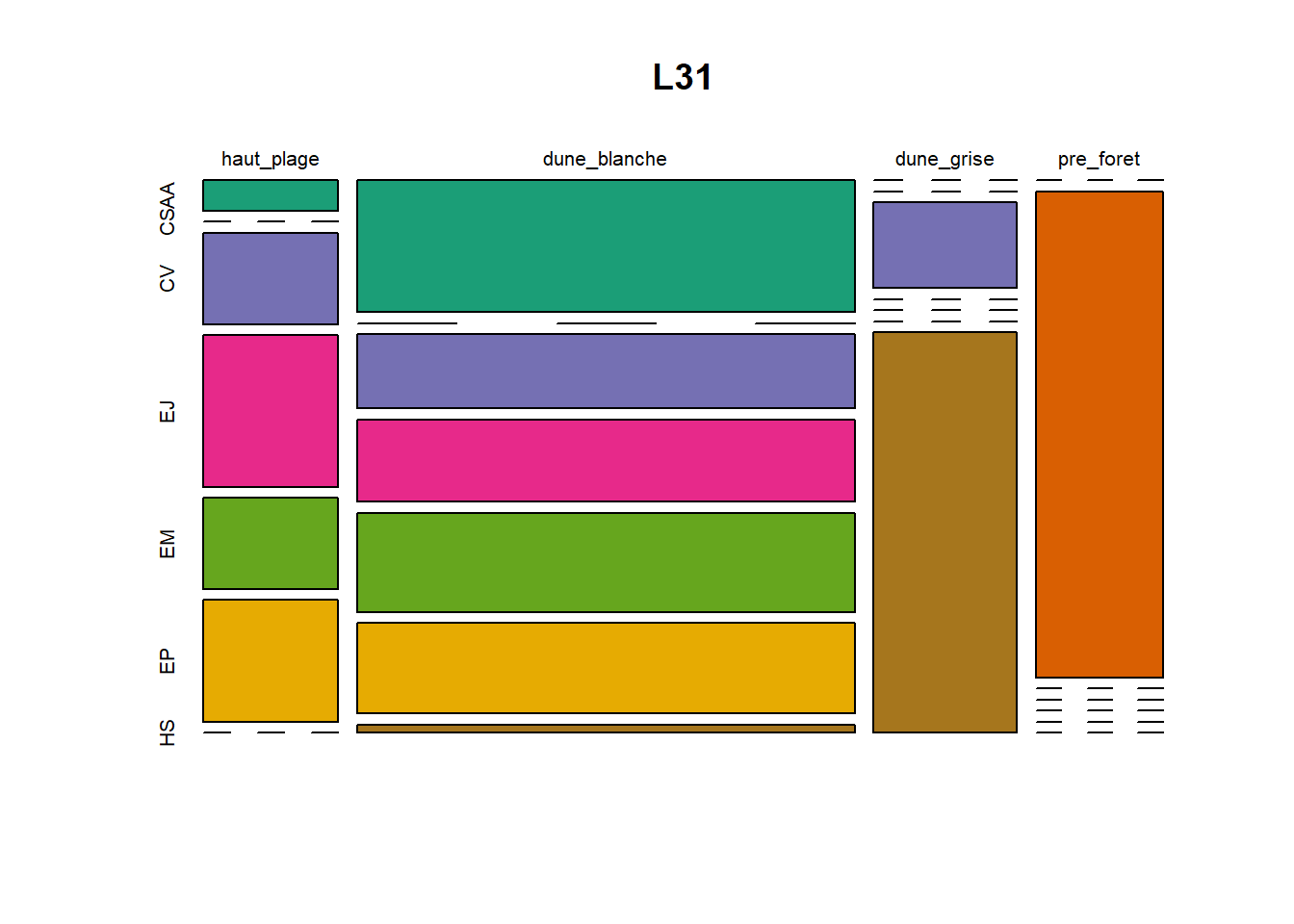
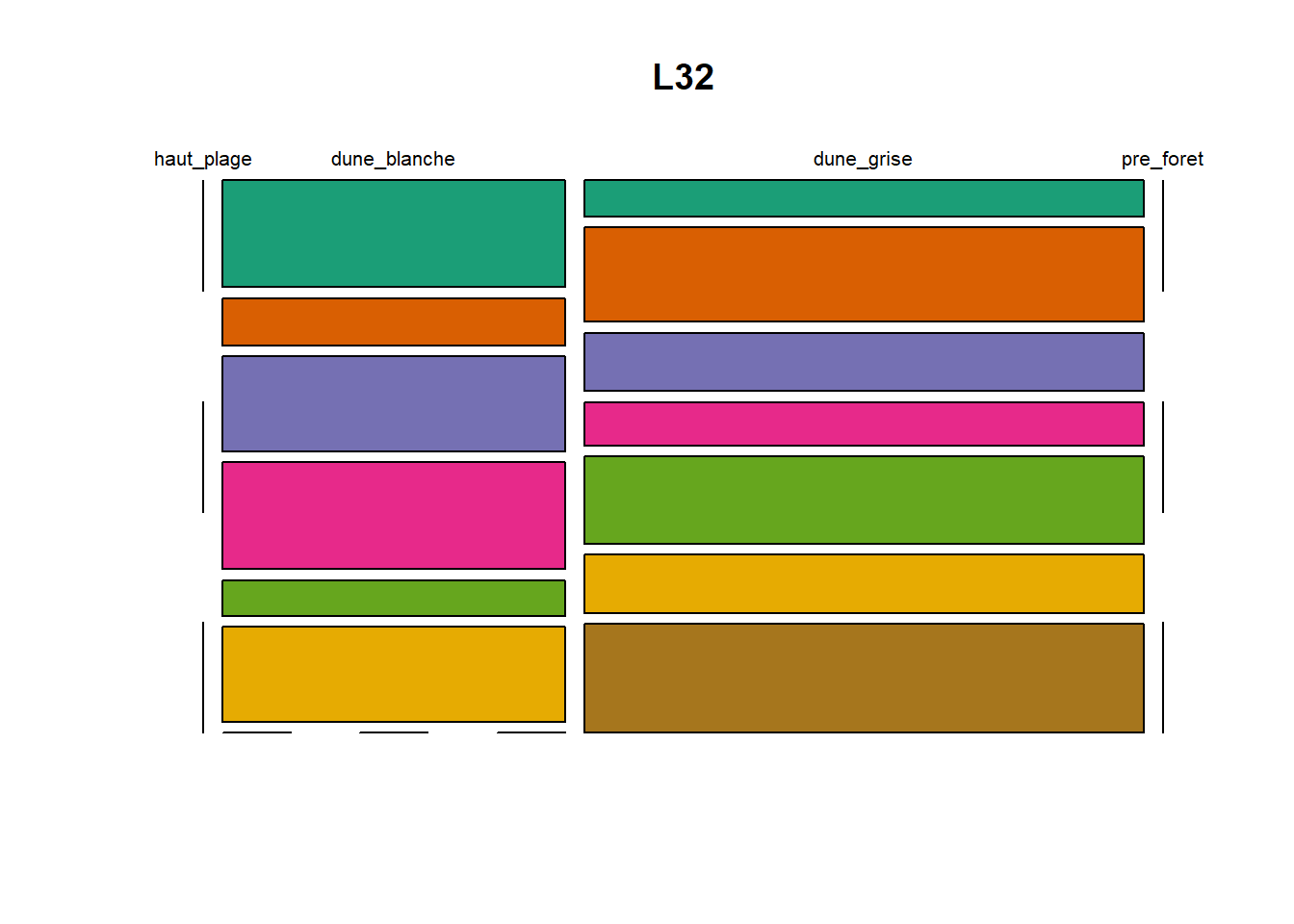
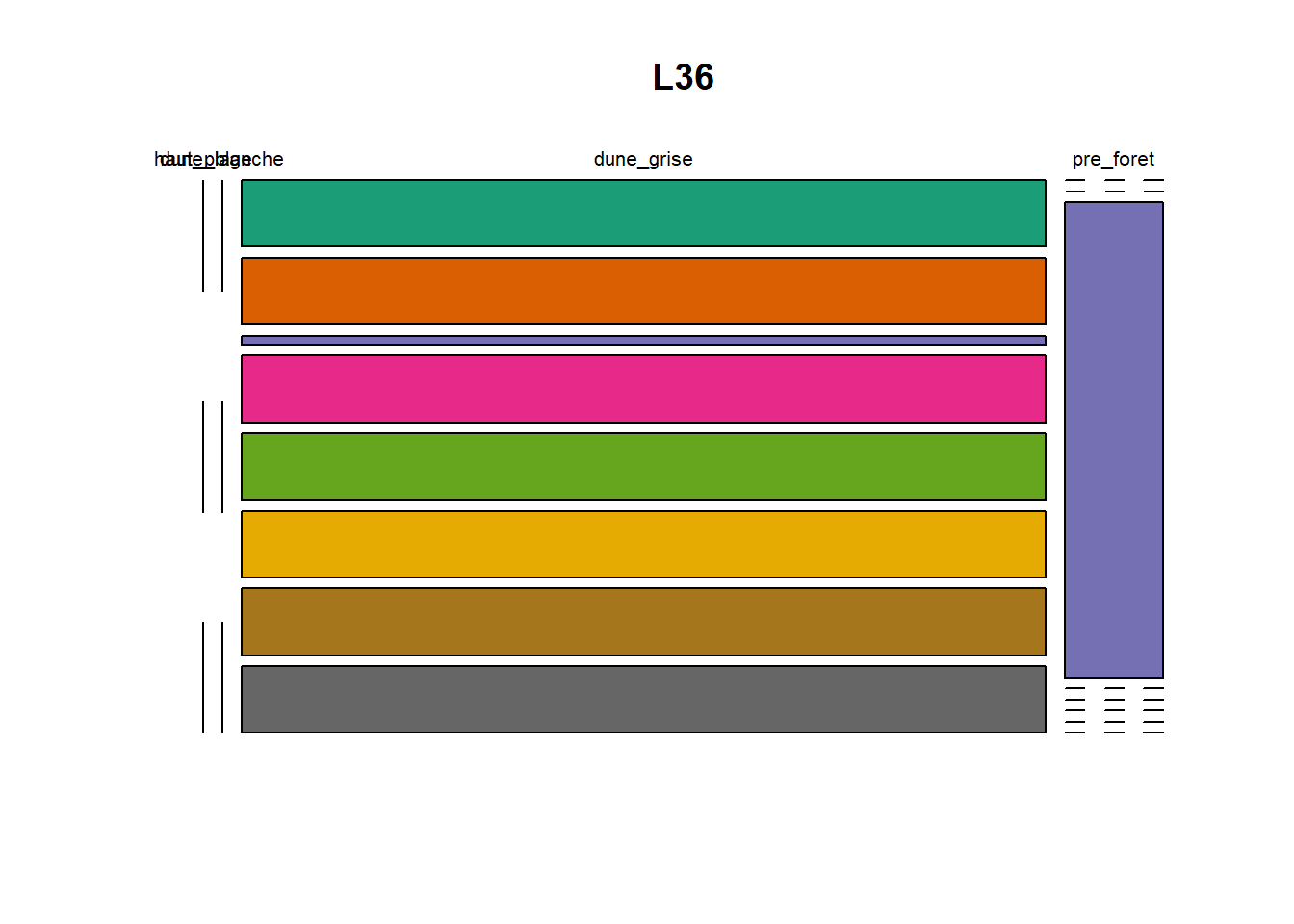
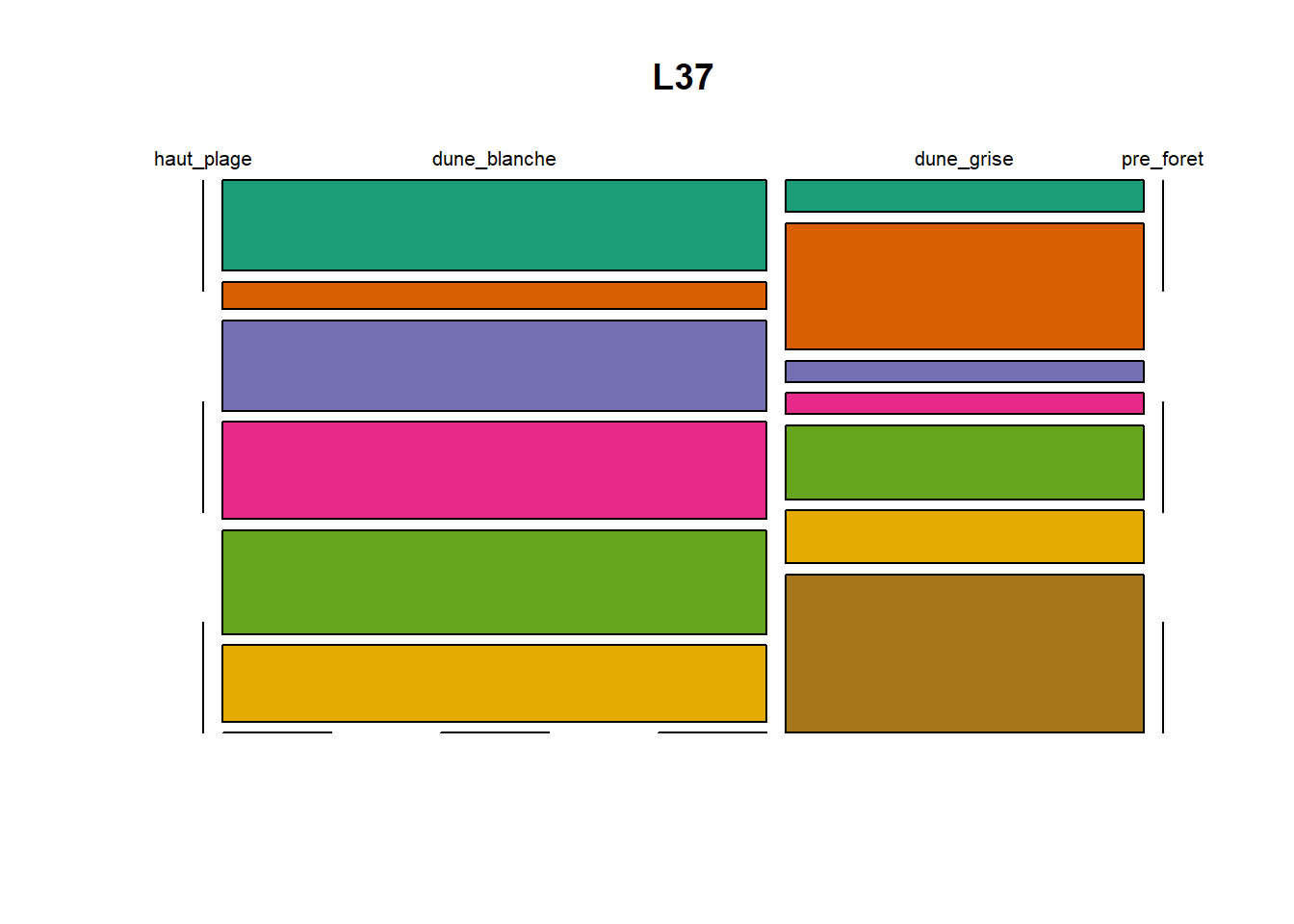
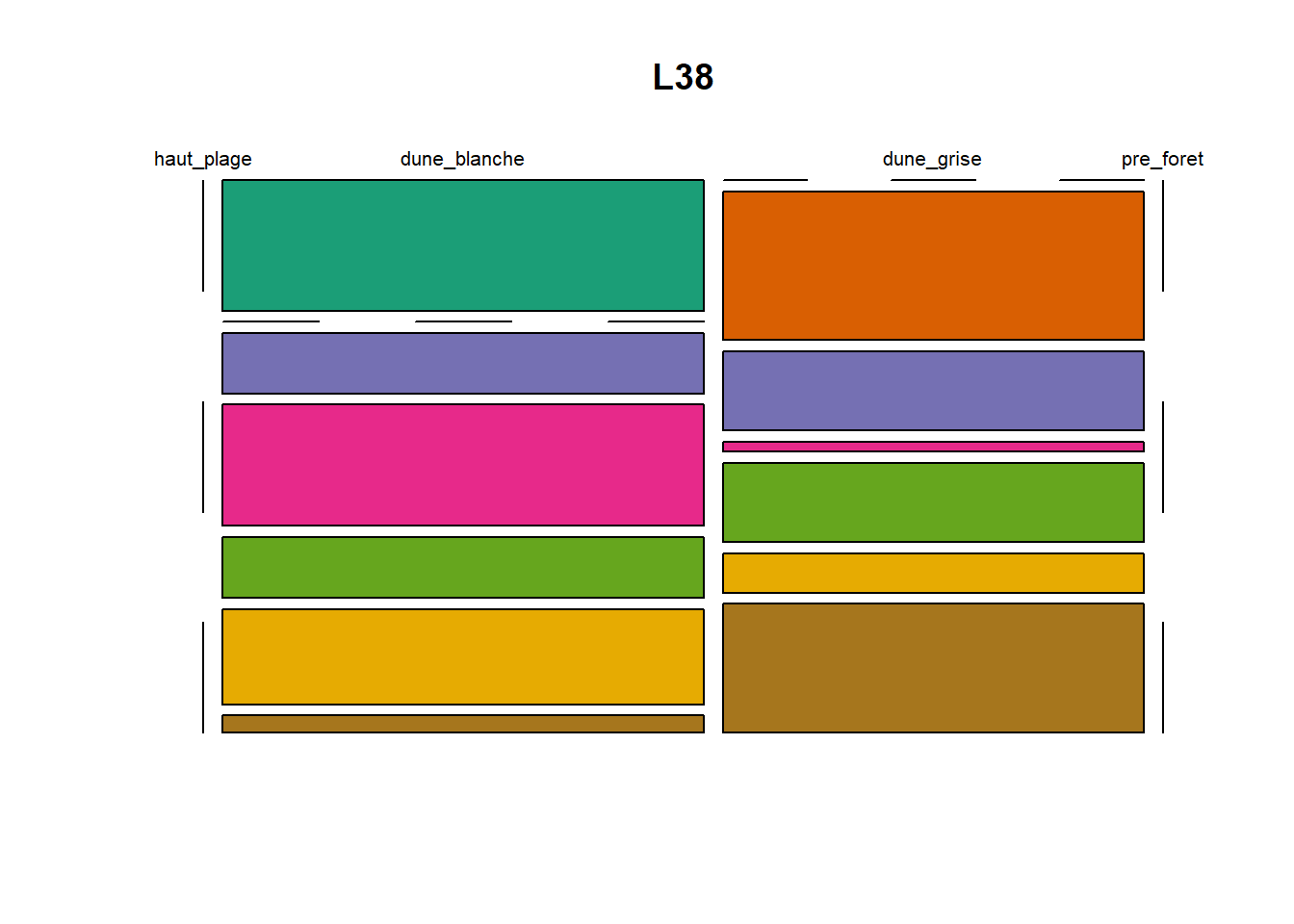
OUTPUT
- specom [8 x 4] : résume le nombre d’individus par espèces par faciès.
- specRecAll [214 x 6] : nombre d’individus par espècse par faciès avec la précision di sute et de nouveau l’espèce + rendu graphique.
Transformation du tableau de génotypes multilocus de chaque espèce en objet genind, en gérant automatiquement la ploïdie (2/4/6), puis en genpop, afin de calculer les F-statistiques de Weir & Cockrham (wc) avec hierfstat. Cett eétape est encapsulé dans la fonction load_genetics, ce qui permet de traiter l’ensemble des espèces de façon homogène.
- Pour l’utiliser individuellement, il suffit d’écrire objet <- load_genetics("i") où i correspond au nom de l’espèce.
- Pour une seule utilisation, il suffit de suivre le script ci-après.
Les F-statistiques de Weir & Cockerham (FST, FIS) ont été calculée avec la fonction hierfstat::wc() utilisable que pour les jeux de données haploïdes ou diploïdes. Par conséquent, ces indices n’ont été estimés que pour les espèces diploïdes de notre jeu de données (CS, CV, EM et EP).
genos <- setNames(
lapply(c("AA", "AC", "CS", "CV", "EJ", "EM", "EP", "HS"), load_genetics),
c("AA", "AC", "CS", "CV", "EJ", "EM", "EP", "HS")
)fst_table <- data.frame(
Ploidy = sapply(genos, `[[`, "ploidy"),
N_ind = sapply(genos, `[[`, "nind"),
N_loc = sapply(genos, `[[`, "nloc"),
FST = round(sapply(genos, `[[`, "FST"), digits = 3),
FIS = round(sapply(genos, `[[`, "FIS"), digits = 3)
)Pour les espèces diploïdes uniquement, exploration de la structuration de la diversitégénétiques à différents niveaux spatiaux (région, communauté, région/communauté imbriquées) à l’aide des F-statistiques hiérarchiques de Weir & Cockerham. Calcul des indices de diversité de Hill afin d’obtenir une mesure synthétique de la diversité génétique par espèce et par niveau.
esp_diploides <- c("CS", "CV", "EM", "EP")
res_all_fst <- lapply(esp_diploides, analyse_espece_fst)
names(res_all_fst) <- esp_diploides
fst_hier_table <- do.call(
rbind,
lapply(names(res_all_fst), function(sp) {
r <- res_all_fst[[sp]]
data.frame(
Espece = sp,
Niveau = c("region", "community", "region/community"),
FST = round(
c(
r$Fst_region$FST,
r$Fst_community$FST,
r$Fst_region_community$FST
),
digits = 3
),
FIS = round(
c(
r$Fst_region$FIS,
r$Fst_community$FIS,
r$Fst_region_community$FIS
),
digits = 3
)
)
})
)OUTPUT
- fst_hier-table : pour chaque espèce diploïde, FST et FIS estimés entre régions, entre communautés et pour une strcture imbriquée région/communauté.
On a déjà genos, un objet contenant les données génétiques par espèces.
Package utilisé
- hillR
AllDiv <- compute_all_hill(esp_diploides)
Hill_gen <- AllDiv %>%
tidyr::pivot_wider(
names_from = div,
values_from = val
) %>%
rename(transect = pop)
# permet de mettre sur le même échelle que la diversité spéciifique (1 valeur par transect)
# moyenner les 9 espèces par transect
Hill_gen_transect <- Hill_gen %>%
group_by(transect) %>%
summarise(
q0_gen = mean(q0, na.rm = TRUE),
q1_gen = mean(q1, na.rm = TRUE),
q2_gen = mean(q2, na.rm = TRUE)
)OUTPUT
- AllDiv : pour chaque espèce diploïde, les indices de diversité de Hill d’ordre 0, 1 et 2.
Je me suis rendue compte que les nombres de Hill ont été calculés uniquement pour les espèces diploïdes. Voici une version avec toutes les espèces.
Package utilisé
- hillR
especes_all <- c("AA", "AC", "CS", "CV", "EJ", "EM", "EP", "HS")
AllDiv_all <- compute_all_hill(especes_all)
Hill_gen_all <- AllDiv_all %>%
tidyr::pivot_wider(
names_from = div,
values_from = val
) %>%
rename(transect = pop)
# permet de mettre sur le même échelle que la diversité spéciifique (1 valeur par transect)
# moyenner les 9 espèces par transect
Hill_gen_transect_all <- Hill_gen_all %>%
group_by(transect) %>%
summarise(
q0_gen = mean(q0, na.rm = TRUE),
q1_gen = mean(q1, na.rm = TRUE),
q2_gen = mean(q2, na.rm = TRUE)
)OUTPUT
- AllDiv : pour chaque espèce diploïde, les indices de diversité de Hill d’ordre 0, 1 et 2.